Sobre as indicações da biópsia renal/estudo histopatológico (importante para avaliar a actividade e potencial reversibilidade das lesões), sugere-se a consulta do capítulo sobre GNA pós-infecciosa.
A avaliação histopatológica pode basear-se na microscopia óptica, na imunofluorescência e na microscopia electrónica.
Apesar da limitação diagnóstica da microscopia óptica, a informação obtida é importante porque permite relacionar as alterações histológicas com a clínica e o prognóstico. A presença de crescentes aponta para a existência de doença grave, geralmente associada a glomerulonefrite rapidamente progressiva. A microscopia óptica também pode esclarecer se a doença glomerular é focal (< 50% dos glomérulos envolvidos) ou generalizada.
A imunofluorescência determina a natureza do processo imune causador da glomerulonefrite. Detecta a deposição de anticorpos e componentes do complemento.
A microscopia electrónica fornece informação sobre a ultraestrutura glomerular, avalia a localização dos imunodepósitos electrodensos e o grau de lesão das células glomerulares.
Na prática clínica importa salientar as seguintes noções:
- Para avaliar a gravidade importa investigar a função renal, quantificar a proteinúria e detectar o mais precocemente possível aspectos histológicos pejorativos como necrose, formação de “meias-luas ou crescentes” e sinais de compromisso intersticial;
- São considerados factores preditivos de reversibilidade das lesões o tamanho dos rins segundo critérios ecográficos e a normalidade da função renal;
- Rins pequenos e hiperecogénicos, associados a hipofunção renal orientam para a probabilidade de resposta deficiente a tratamento com corticóides ou imunossupressores;
- Rins de tamanho normal associados a função renal normal ou minimamente reduzida são muito provavelmente preditivos de boa resposta ao tratamento, especialmente se a diurese estiver conservada.
A avaliação histopatológica pode basear-se na microscopia óptica, na imunofluorescência e na microscopia electrónica.
Apesar da limitação diagnóstica da microscopia óptica, a informação obtida é importante porque permite relacionar as alterações histológicas com a clínica e o prognóstico. A presença de crescentes aponta para a existência de doença grave, geralmente associada a glomerulonefrite rapidamente progressiva. A microscopia óptica também pode esclarecer se a doença glomerular é focal (< 50% dos glomérulos envolvidos) ou generalizada.
A imunofluorescência determina a natureza do processo imune causador da glomerulonefrite. Detecta a deposição de anticorpos e componentes do complemento.
A microscopia electrónica fornece informação sobre a ultraestrutura glomerular, avalia a localização dos imunodepósitos electrodensos e o grau de lesão das células glomerulares.
Aspectos gerais do tratamento
Importa salientar que em grande número de GN as medidas terapêuticas são inespecíficas reservando-se os imunossupressores para as formas mais agressivas.
Constituem factores de mau prognóstico o sexo masculino, a maior idade no início do quadro clínico, a proteinúria de tipo nefrótico e a presença de fibrose intersticial como achado histopatológico renal.
Os imunossupressores ocupam um lugar importante no tratamento de glomerulonefrites com mecanismo autoimune subjacente.
Os tratamentos biológicos mais recentes, seletivos e menos tóxicos, como a depleção dos linfócitos B e inibição do complemento começam a encontrar o seu uso clínico em várias formas de glomerulonefrite.
ENTIDADES ESPECÍFICAS
NEFROPATIA por IgA
Importância do problema
A nefropatia por IgA, também designada por nefropatia de Berger, é a glomerulonefrite primária mais frequente a nível mundial. Trata-se duma afecção responsável por insuficiência renal terminal em 20 a 50% dos adultos e em 2,5 a 9% das crianças. Surgindo em todas as idades, mas principalmente na segunda e terceira décadas, predomina no sexo masculino (M:F de 2:1 a 6:1), raramente ocorrendo na raça negra. A incidência geográfica varia muito (18-40% no Japão e 2-10% no Canadá, Estados Unidos e Grã-Bretanha).
Embora seja classicamente considerada uma doença esporádica, salienta-se que existem formas familiares.
Etiopatogénese
A nefropatia por IgA é caracterizada por proliferação mesangial com depósitos de IgA (subtipo IgA1) nesta área, na ausência de doença sistémica. Por vezes podem observar-se depósitos de IgG, IgM e C3 no mesmo local com menor intensidade.
A associação de episódios de hematúria macroscópica a infecções das vias respiratórias superiores sugere que esta resposta imune seja induzida por antigénios microbianos.
Embora se admita que os mecanismos de lesão glomerular sejam específicos, admite-se a intervenção de factores hemodinâmicos e vasculares como o sistema endotélio-monóxido de azoto, citocinas e os factores de crescimento como a interleucina 6 e o factor de crescimento das plaquetas, entre outros.
Manifestações clínicas
A nefropatia por IgA tem apresentação clínica, evolução e prognóstico muito variáveis. Em países que aplicam programas de rastreio nas escolas como o Japão, a nefropatia por IgA é mais frequentemente diagnosticada na sua fase inicial pela detecção de hematúria microscópica assintomática.
Classicamente descrevem-se as seguintes formas de apresentação: – surtos com duração em geral inferior a uma semana de hematúria macroscópica; surtos de micro-hematúria e/ou proteinúria assintomáticas; – síndroma nefrítica aguda; – síndroma nefrótica; e – síndroma nefrítica associada a síndroma nefrótica.
A forma de apresentação mais frequente é a hematúria macroscópica recorrente que habitualmente surge em simultâneo ou 2 a 3 dias após o início de infecção das vias respiratórias superiores. Os intervalos inter-surtos são variáveis, sendo que a segunda forma de apresentação mais frequente é a micro-hematúria assintomática com ou sem proteinúria.
Diagnóstico
Não existem marcadores serológicos ou urinários específicos para o diagnóstico de nefropatia por IgA. Os valores de IgA não têm valor diagnóstico porque estão elevados em apenas 15% dos doentes pediátricos. Os valores do complemento (C3 e C4) estão normais.
Para diagnóstico diferencial com a GNA pós-infecciosa deve atender-se ao facto de na nefropatia por IgA nunca se encontrar intervalo livre (ou o intervalo livre é muito menor, 2-3 dias) entre a infecção e o aparecimento da hematúria.
Para estabelecer o diagnóstico definitivo é necessário praticar a biópsia renal. As formas de síndroma nefrítica-nefrótica associadas correspondem a histopatologia com lesões glomerulares graves e prognóstico mais reservado com evolução altamente provável para insuficiência renal terminal (IRT).
Tratamento
Actualmente não existe tratamento curativo para a nefropatia por IgA. Como actuação prática está indicado o controlo da pressão arterial e a administração de inibidores da enzima de conversão da angiotensina ou de antagonistas dos receptores da angiotensina II. Estes fármacos são efectivos na redução da proteinúria e atrasam a progressão da doença glomerular.
Prognóstico
Na idade pediátrica a progressão para doença renal crónica terminal (na proporção de 10% no prazo de 20 anos) é mais lenta que nos adultos. Nalguns casos foi descrita remissão espontânea. A verificação de proteinúria prolongada do tipo nefrótico, hipofunção renal e HTA são factores de mau prognóstico.
Seguidamente são abordadas determinadas entidades específicas englobadas no conceito de Doença Glomelurar.
NEFROPATIA na PÚRPURA de HENOCH-SCHONLEIN
Definição e importância do problema
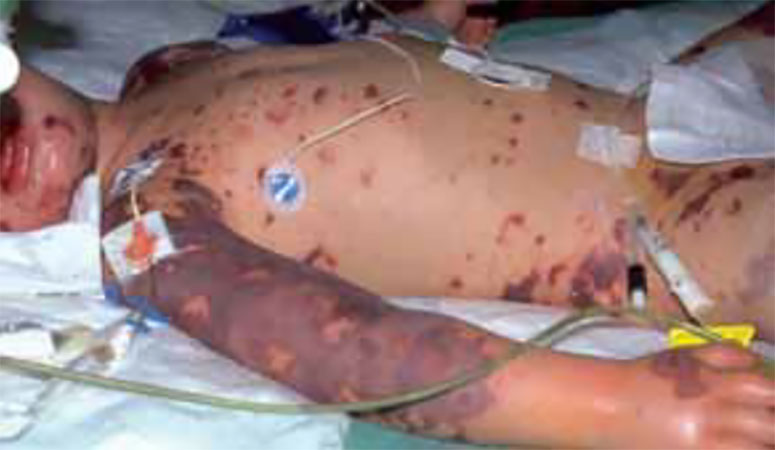
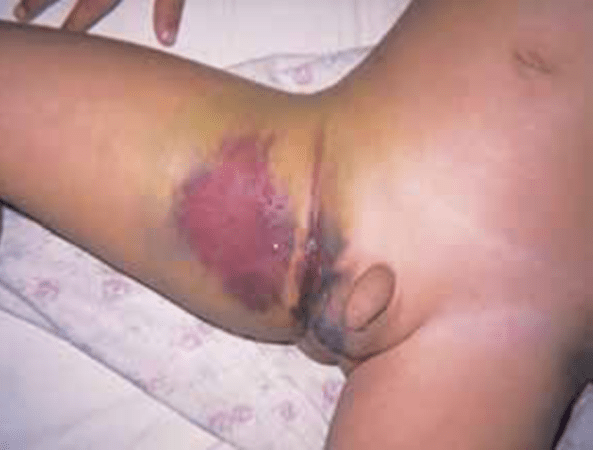
A púrpura de Henoch-Schӧnlein (PHS), um exemplo de nefropatia associada a doença sistémica, é uma vasculite leucocitoclástica de pequenos vasos caracterizada por púrpura cutânea, artralgia, dor abdominal e nefrite. Aproximadamente em 20 a 80% dos doentes com PHS surgem manifestações renais que variam desde hematúria microscópica assintomática a glomerulonefrite progressiva e grave.
Etiopatogénese
Sob o ponto de vista da histopatologia renal, o quadro é sobreponível ao descrito para a nefropatia por IgA: deposição de IgA polimérica nos pequenos vasos do glomérulo e na parede dos pequenos vasos a nível sistémico.
Existe produção aumentada de IgA1 aberrante, deficiente em galactose (Gd-IgA1). As moléculas de Gd-IgA1, reconhecidas por anticorpos IgA e IgG, formam imunocomplexos (IC). Estes depositam-se no mesângio e podem desencadear lesão no glomérulo. As células mesangiais começando a proliferar com sobreprodução de matriz extracelular e citocinas, inicia-se um processo de inflamação glomerular e ulterior esclerose. Pode verificar-se a presença de crescentes.
A lesão renal característica consiste, pois, numa glomerulonefrite proliferativa mesangial com depósitos de IgA granular no mesângio, de distribuição focal ou segmentar. Pode também ser observada lesão tubulointersticial com fibrose, atrofia e infiltração celular linfocitária.
Manifestações clínicas
A PHS afectando todas as idades, em 90% dos casos ocorre antes dos 10 anos com uma mediana de idade de 6 anos. Há predomínio ligeiro do sexo masculino.
A apresentação clínica clássica é o aparecimento da púrpura cutânea palpável, com distribuição simétrica e de predomínio nos membros inferiores, na ausência de trombocitopénia ou de alteração da coagulação. Segue-se o atingimento articular em forma de artralgia e artrite de predomínio nas articulações dos joelhos e tornozelos, assim como o atingimento digestivo com dor abdominal que pode acompanhar-se de hemorragia digestiva.
As manifestações da nefrite associada a PHS estão presentes em 90% dos doentes oito semanas após o episódio inaugural de PHS. A forma de apresentação mais comum é a hematúria macroscópica; contudo, menos frequentemente podem surgir outros quadros: proteinúria nefrótica/não nefrótica, HTA, síndroma nefrótica, síndroma nefrítica e a combinação destas últimas.
Numa pequena proporção de casos pode surgir insuficiência renal.
Tratamento
O tratamento do episódio agudo da PHS é sintomático com repouso relativo, hidratação e analgesia.
Nos casos de nefropatia moderada ou grave, decorrendo dos resultados da biópsia renal poderão estar indicados corticóides, imunossupressores e inibidores da enzima de conversão da angiotensina ou de antagonistas dos receptores da angiotensina II.
Prognóstico
A nefropatia associada a PHS na maioria dos doentes tem um excelente prognóstico. A resolução completa e espontânea da afecção ocorre habitualmente nos doentes com manifestações renais ligeiras. Há factores de risco conhecidos que podem indiciar evolução menos favorável da nefrite na PHS: HTA no início, alteração da função renal no momento do diagnóstico, proteinúria nefrótica persistente e achado histopatológico de crescentes.
No entanto, o desenvolvimento de lesão renal ao longo do tempo não é previsível, podendo inclusivamente acontecer em contexto de manifestações iniciais ligeiras.
NEFROPATIA LÚPICA
Definição e importância do problema
O lúpus eritematoso sistémico (LES) é uma doença autoimune, multissistémica, de etiologia desconhecida, com formação de imunocomplexos provocando lesão tecidual do rim. A maior parte dos depósitos contém IgG e C3; por vezes, também IgM ou IgA. Os respectivos autoanticorpos actuam predominantemente contra antigénios nucleares.
Atinge sobretudo as mulheres jovens e somente 10 a 17% dos casos são diagnosticados antes dos 16 anos. (ver capítulo sobre LES na Parte sobre Reumatologia)
A nefropatia (glomerulonefrite) é uma complicação frequente do LES, surgindo em 30 a 80% dos doentes pediátricos. Rara antes dos 5 anos, atinge em particular o grupo etário acima dos 10 anos, e em apenas 25% destes doentes a doença renal surge como primeira manifestação.
A biópsia renal e a avaliação histopatológica, constituindo o critério padrão de ouro/gold standard para estabelecer o diagnóstico de nefropatia lúpica, são importantes para a orientação do regime terapêutico a instituir.
Histopatologia
De acordo com a classificação da nefropatia lúpica da Organização Mundial da Saúde (OMS), baseada na combinação dos achados histopatológicos na microscopia óptica (MO), imunofluorescência (IF) e microscopia electrónica (ME), foram descritas seis classes de alterações histológicas:
- Classe I: glomerulonefrite (GN) lúpica mesangial mínima, não apresentando alterações histológicas na MO; através da IF e da ME detectam-se depósitos imunes mesangiais;
- Classe II: GN proliferativa mesangial: a MO revela hipercelularidade mesangial e aumento da matriz com depósitos mesangiais contendo imunoglobulinas e complemento;
- Classe III: GN com lesões mesangiais e endocapilares em < 50% dos glomérulos;
- Classe IV: GN com lesões em ≥ 50% de glomérulos atingidos;
- Numa subclassificação gradua-se a lesão proliferativa em: *segmentar se < 50% do tufo glomerular estiver envolvido, e *global se ≥ 50% do tufo glomerular estiver afectado;
- Classe V: GN lúpica membranosa;
- Classe VI: GN com glomérulos globalmente esclerosados (> 90% dos glomérulos com esclerose), fibrose intersticial e atrofia tubular sem sinais de actividade imunológica.
É possível a evolução de um padrão histológico para outro de maior gravidade, o que se poderá relacionar com tratamento inadequado.
Manifestações clínicas
A nefropatia lúpica manifesta-se de modo diverso, variando desde a ausência de sinais ou sintomas, até à presença de proteinúria (< 1 g/24 horas), hematúria, hipertensão arterial e insuficiência renal, isoladamente ou em associação. A verificação de um sedimento urinário rico em elementos celulares, com eritrócitos, leucócitos, cilindros eritrocitários, granulosos ou de leucócitos e cilindros grossos e granulosos é muito sugestiva de LES; de referir no entanto, que tais características também podem surgir noutras conectivites.
A proteinúria é o achado mais frequente, muitas vezes precedendo o aparecimento de síndroma nefrótica.
Não há relação directa entre as manifestações clínicas do LES e o grau de atingimento renal.
Exames laboratoriais
O diagnóstico é sugerido também pela detecção de autoanticorpos circulantes (antinucleares, anti-DNA, antinucleossomas e anti a-actinina, e avaliação sérica de C3 e C4.
A actividade da doença é monitorizada, não só pela clínica, mas também pela evolução destes parâmetros, e pela avaliação da função renal, da velocidade de sedimentação e das alterações urinárias. Na maior parte dos casos de doença activa os valores séricos de C3 e C4 estão diminuídos.
Tratamento
A precocidade do início da terapêutica é muito importante: permite reduzir o número de recorrências, a frequência de evolução para a insuficiência renal, assim como a mortalidade. O objectivo é induzir remissão clínica rápida, evitar a progressão da doença renal, evitar recaídas com o mínimo possível de efeitos colaterais, designadamente relacionados com a toxicidade farmacológica.
A escolha do esquema terapêutico é um desafio devido à heterogeneidade da doença, à apresentação clínica e à evolução, imprevisível.
Existe uma grande variedade de opções terapêuticas (gerando controvérsia), sendo que nenhum esquema é totalmente eficaz. A controvérsia diz respeito sobretudo à escolha do agente imunossupressor, (azatioprina ou ciclofosfamida), sendo o uso de corticóides (prednisona) universalmente aceite. Nos doentes com nefrite da classe IV tem sido utilizado o anticorpo monoclonal rituximab.
Prognóstico
O prognóstico da nefropatia lúpica depende do balanço entre a eficácia terapêutica e os possíveis efeitos colaterais do tratamento farmacológico (por ex. risco de HTA, osteoporose, obesidade e diabetes mellitus, associados a corticoterapia crónica).
A sobrevivência dos doentes com nefrite lúpica tem vindo a aumentar muito nos últimos 30 a 40 anos, salientando-se contudo uma mortalidade de cerca de 15 a 20%. A infecção, muitas vezes secundária à imunossupressão, constitui a maior causa de mortalidade. Do mesmo modo a doença cardiovascular associada tem sido progressivamente identificada como causa importante de morbilidade a longo prazo.
A insuficiência renal poderá surgir em cerca de 30% dos doentes.
GLOMERULOESCLEROSE SEGMENTAR e FOCAL (GESF)
Definição e importância do problema
A GESF, entidade clinicopatológica com várias etiologias e mecanismos patogénicos, define-se pela existência duma lesão histopatológica comum: esclerose do glomérulo de forma focal (percentagem variável de glomérulos) e segmentar (uma parte do glomérulo). Na idade pediátrica corresponde a 7 a 15% dos casos de síndroma nefrótica. A GESF pode ser classificada em primária e secundária.
Manifestações clínicas
Esta doença glomerular, na sua forma primária ou idiopática, manifesta-se habitualmente como síndroma nefrótica.
A forma secundária associa-se a proteinúria não nefrótica associada a certo grau de lesão renal. Inclui entidades clínicas com redução de massa renal funcionante (diminuição do número de glomérulos) em que há uma resposta adaptativa traduzida por hipertrofia glomerular e hiperfiltração. Citam-se como exemplos situações com antecedentes de prematuridade ou de restrição de crescimento intrauterino, de infecções ou de tratamentos com certos fármacos.
Estão descritas formas familiares de GESF resultando de mutações de genes codificadores de proteínas dos podócitos (proteinúria familiar).
Tratamento e prognóstico
O tratamento das formas secundárias é o da doença de base.
O tratamento da GESF primária engloba tratamento conservador e imunossupressão para controlar a proteinúria e preservar a função renal. Inclui as medidas gerais do tratamento das GN utilizando IECA ou ARA II com o objectivo de redução da proteinúria e de tratamento da HTA para retardar a progressão da doença renal.
Sendo a síndroma nefrótica a apresentação nas formas primárias, em tal contexto estão habitualmente indicados os corticóides. Com este tratamento verifica-se remissão em 20 a 25% dos doentes.
Nos doentes evidenciando corticorresistência e em que a biópsia estabelece o diagnóstico de GESF, existem varias modalidades terapêuticas com imunossupressores.
Em suma, a eficácia do tratamento varia em função da história natural da doença. As formas primárias e do foro genético tendem a evoluir para doença renal crónica terminal. Nos casos evoluindo com proteinúria não nefrótica a evolução é mais favorável.
NEFROPATIA MEMBRANOSA
Definição e etiopatogénese
A nefropatia membranosa é uma entidade clinicopatológica bem definida caracterizada por espessamento difuso e homogéneo da membrana basal glomerular (MBG) e depósitos granulares difusos de IgG e C3 (imunocomplexos) de localização subepitelial, que desenham o contorno das paredes capilares; trata-se de reacção de autoanticorpos contra antigénios dos podócitos.
São considerados vários estádios ou graus, de I a V, em função da topografia dos depósitos, da espessura da MBG e do grau de esclerose glomerular.
Os nefrologistas salientam que o termo de nefropatia é preferível ao de glomerulopatia porque não se verificam sinais inflamatórios ao nível do glomérulo.
Na maioria das vezes idiopática, pode contudo ser secundária a infecções (hepatites B e C), fármacos, LES, artrite reumatóide, tumores, etc..
Manifestações clínicas
A manifestação clínica mais habitual (~40-75% dos casos) da nefropatia membranosa é a síndroma nefrótica; menos frequentemente, proteinúria assintomática. É frequente, também, hematúria microscópica, e rara, a hematúria macroscópica. O fenómeno de trombose venosa, nomeadamente da veia renal, no contexto de síndroma nefrótica, é mais frequente na nefropatia. O diagnóstico é estabelecido por biópsia renal.
Tratamento e prognóstico
Nos doentes com proteinúria ligeira e função renal normal a terapêutica recomendada é conservadora. Com efeitos, em cerca de 60% dos doentes verifica-se remissão expontânea. Em função do contexto clínico de cada caso, poderão ser utilizados inibidores da enzima de conversão da angiotensina ou antagonistas dos receptores da angiotensina II.
Nos doentes com síndroma nefrótica e/ou diminuição da função renal a recomendação terapêutica inclui os fármacos anteriores e imunossupressores.
GLOMERULONEFRITE (GN) MEDIADA por C3/GNC3 e GN MEDIADA por IMUNOCOMPLEXOS (IC)/GNIC
Definição e importância do problema
Também denominada glomerulonefrite membranoproliferativa (GNMP), ou mesangiocapilar, a GNC3 e GNIC é uma glomerulopatia de etiopatogénese não completamente esclarecida, caracterizada por proliferação de células mesangiais e incremento da matriz mesangial, espessamento difuso da parede capilar glomerular, do que resulta aspecto lobular do glomérulo.
Admite-se uma incidência, rara, de 1-2 casos por milhão, sendo excepcional o surgimento antes dos 5 anos de idade.
Etiopatogénese
A nova classificação baseia-se mais no mecanismo fisiopatológico do que nas características histopatológicas. Na GNC3 a desregulação da via alternativa do complemento leva a deposição predominante de C3, o que a diferencia da GNIC, em que se verifica predomínio de deposição de IgG (e por vezes de IgM e IgA) detectada por imunofluorescência.
A microscopia electrónica permite subdividir a GNC3 em duas formas: glomerulonefrite C3, e doença dos depósitos densos (DDD), de acordo com o padrão de distribuição dos depósitos imunológicos.
A GNC3 é considerada uma doença primária do complemento em que a deposição deste resulta de um defeito na via alternativa do complemento. Mutações ou autoanticorpos que afectam os activadores ou reguladores desta via do complemento, em particular a alteração da C3 convertase (factor nefrítico C3- C3NEF) foram detectados em > 80% dos doentes com GNC3. Na GN-IC também há activação do complemento, mas através da via clássica.
Os achados da histopatologia traduzem-se por deposição de imunoglobulinas/imunocomplexos e/ou proteínas do complemento no mesângio e/ou nas paredes dos capilares do glomérulo.
De acordo com a localização dos depósitos de IC, são descritos três tipos- I, II e III, não identificados por microscopia óptica.
Manifestações clínicas e laboratoriais
Na idade pediátrica predomina a forma idiopática. As formas secundárias manifestam-se na sequência de infecções crónicas, doenças imunitárias, neoplasias, entre outras.
Os doentes podem apresentar: proteinúria, com ou sem síndroma nefrótica, hematúria e síndroma nefrítica com atingimento importante da função renal. A HTA está presente em 60 a 80% dos doentes.
Em 80 a 90% dos casos existe hipocomplementémia persistente (com diminuição de C3 e/ou C4 em função da via de ativação do complemento atingida).
Nível baixo de C3 é sugestivo de GNC3, especialmente na ausência de história sugestiva de glomerulonefrite pós-infecciosa.
O diagnóstico definitivo é feito pela biópsia renal com o estudo da microscopia óptica, imunofluorescência e microscopia electrónica.
É importante estabelecer o diagnóstico diferencial da GNMP (GNC3/GNIC) com GN pós-estreptocócica/GNPE).
Devem ser valorizados os seguintes dados clínicos: – Idade (GNPE*à 3-12 anos; GNMP**àadolescência); – Edema (*à por retenção de fluidos; **à secundário a síndroma nefrótica); – HTA (*à transitória; **à persistente); -Albumina sérica ( *à normal ou ligeira diminuição; **à moderadamente ou muito diminuída); – Proteinúria (*à mínima ou moderada transitória; **à moderada ou maciça persistente); – Função renal (*à normal ou reduzida transitória; **à normal ou reduzida persistente); – C3 (*à baixo, normalizando em 6-8 semanas; **à baixo persistente).
Tratamento
No tratamento da forma primária os imunossupressores são uma arma com eficácia ainda não totalmente comprovada. São utilizados nos casos de síndroma nefrótica com/sem diminuição da função renal. Nas formas ligeiras com proteinúria moderada e função renal normal e estável, os IECA e ARA II estão indicados. Importa igualmente o tratamento da HTA. A detecção do factor nefrítico C3 (C3NEF) pode estabelecer a indicação de tratamento com rituximab, um anticorpo monoclonal anti-CD20.
Prognóstico
A GNMP é uma das doenças glomerulares com prognóstico mais reservado uma vez que, após 10-15 anos de seguimento, em cerca de 50% dos pacientes surge falência renal terminal, proporção que atinge 80-90% após 20 anos de evolução. As remissões são raras.
NEFROPATIA DIABÉTICA
Definição e importância do problema
A doença renal na diabetes (DRD) constitui uma das complicações graves da doença. De acordo com a história natural da doença, podem ser identificados cinco estádios ao longo da idade: estádios I e II (nefropatia pré-clínica) caracterizados por hiperfiltração glomerular- inicial; estádio III (nefropatia incipiente) caracterizado por normoalbuminúria, embora com lesões estruturais demonstradas por biópsia- 5 anos; estádio IV (nefropatia clínica) em que existe microalbuminúria- 10 anos; estádio V (nefropatia terminal) caracterizado por proteinúria, HTA e deterioração da filtração glomerular- 20 anos.
A DRD, que também surge na diabetes do tipo 2, tem uma história natural diferente; as respectivas lesões estruturais renais são mais heterogéneas.
Etiopatogénese
Os mecanismos de início e progressão da doença não estão ainda completamente esclarecidos. Admite-se certa predisposição genética associada a factores metabólicos (desregulação glicémica) e hemodinâmicos (hiperfiltração glomerular). Outros factores descritos com importância na patogénese incluem citocinas e factores de crescimento endotelial.
A lesão característica da nefropatia diabética é a expansão mesangial contribuindo para reduzir o lume capilar e, portanto, a área de filtração glomerular. Verifica-se igualmente espessamento da membrana basal glomerular, permitindo a passagem de proteínas de carga negativa, como a albumina.
No estádio avançado de DRD verifica-se expansão mesangial marcada, espessamento da membrana basal, fibrose intersticial, atrofia tubular e glomeruloesclerose.
Manifestações clínicas e laboratoriais
Nos estádios precoces (I a III) os sinais clínicos são subtis, tais como alteração do ritmo circadiano da pressão arterial, designadamente com elevação dos valores durante a noite e microalbuminúria.
A microalbuminúria (MA), definida como excreção de 30-299 mg/dia de albumina ou relação albumina/creatinina de 30-299 mg/g em, pelo menos, duas de três amostras, é um sinal precoce de DRD ocorrendo em 26% das crianças e adolescentes após 10 anos e em 51% após 19 anos de diabetes. Na apresentação clássica de DRD, quando surge MA, a excreção de albumina continua a elevar-se, particularmente na presença de outros factores de risco.
Nas fases mais avançadas (IV e V), verifica-se proteinúria, HTA e compromisso da filtração glomerular.
Nem todos os doentes com microalbuminúria evoluem inexoravelmente para estádios mais avançados de DRD. A MA pode representar lesão endotelial reversível e a regressão para normoalbuminúria é possível se houver um bom controlo glicémico e da pressão arterial mas é independente da inibição do sistema renina-angiotensina.
Diagnóstico e actuação prática
Desde o início da diabetes deve proceder-se à avaliação seriada da microalbuminúria. Se for negativa, deverá repetir-se a avaliação anualmente; se for positiva e persistente, estão indicadas medidas terapêuticas de renoprotecção com o objectivo de prevenir a progressão da lesão renal. Uma das medidas é proceder à monitorização ambulatória da pressão arterial durante 24 horas para detectar precocemente alterações do ritmo circadiano da mesma.
Tratamento
O tratamento engloba a actuação ao nível dos diversos factores de risco da DRD: hiperglicemia, HTA, microalbuminúria e dislipoproteinémia. Os IECA e ARA II são os fármacos de eleição como actuando contra a proteinúria e a HTA. Na idade pediátrica é recomendada a dieta normoproteica.
Prognóstico
Para além da duração da diabetes, constituem factores de risco de agravamento da nefropatia, antecedentes familiares de doença renal, HTA, dislipoproteinémia, índice de massa corporal elevado e tabagismo.
GLOMERULONEFRITE RAPIDAMENTE PROGRESSIVA
Definição e importância do problema
O termo glomerulonefrite rapidamente progressiva (GNRP) refere-se à síndroma clínica resultante de agressão glomerular grave, caracterizada por deterioração aguda e rápida da função renal que, na ausência de tratamento, evolui em poucas semanas ou meses para doença renal terminal.
Pode ocorrer nas diversas formas de glomerulonefrite (GN) primária e secundária. A característica histológica comum é a presença de crescentes (C) na maioria dos glomérulos (GNC). A GNRP pode classificar-se: – de acordo com a alteração imunológica subjacente; ou em: – primária e secundária, como nas outras glomerulonefrites.
Etiopatogénese
O padrão verificado através da imunofluorescência e da microscopia electrónica orienta para o processo etiológico. Assim: – depósitos lineares de IgG indicam glomerulonefrite por anticorpos antimembrana basal; – depósitos granulares de várias imunoglobulinas e/ou fracções de complemento orientam para um processo mediado por imunocomplexos; – a escassez de depósitos imunológicos indica um padrão paucimune típico das glomerulonefrites rapidamente progressivas associadas a ANCA.
A GNRP pode ser dividida de acordo com o processo etiológico subjacente em I, II e III tipos. Doentes com vasculite sistémica apresentam particular tendência para desenvolvimento de GNRP. Doentes com púrpura Henoch-Schӧnlein, poliangeíte microscópica, granulomatose com poliangeíte e LES constituem o maior grupo de doentes com GNRP. (Quadro 4)