Introdução
As coagulopatias congénitas são afecções causadas por alteração dos genes que codificam um ou vários factores implicados na cascata da coagulação. A hemofilia A e a hemofilia B, juntamente com a doença de von Willebrand, integram na sua grande maioria (cerca de 95%) as deficiências congénitas dos factores de coagulação.
As restantes coagulopatias (deficiências de fibrinogénio, factores II, V, VII, X, XI, XIII, assim como as deficiências combinadas de factores V e VIII, pertencem ao grupo das chamadas doenças raras, com uma incidência de 1/500.000 a 2.000.000 nas formas homozigóticas.
O Quadro 1 sintetiza as anomalias básicas da hemostase (tempo de protrombina/TP e tempo de tromboplastina parcial activado/TTPa ou aPTT) relacionadas com os défices dos factores discriminados antes, exceptuando o factor XIII.
QUADRO 1 – Anomalias básicas da hemostase relacionadas com o factor deficiente
| Factor deficiente | TP | TTPa |
| VII | Anormal | Normal |
| VIII, IX e XI | Normal | Anormal |
| Fibrinogénio, II, V, V+VIII e X | Anormal | Anormal |
Definição e importância do problema
A hemofilia é uma coagulopatia congénita causada pelo défice quantitativo de factor VIII (F VIII) da coagulação – hemofilia A, ou de factor IX (F IX) – hemofilia B. Trata-se de doenças monogénicas, resultantes de uma mutação do gene F8 na hemofilia A, e F9 na hemofilia B, ambos localizados no braço longo do cromossoma X (Xq28); portanto, com um padrão de hereditariedade ligada ao sexo.
Nas restantes coagulopatias atrás citadas, a maioria dos genes responsáveis localiza-se nos cromossomas autossómicos, o que lhes confere a característica de hereditariedade mendeliana clássica.
A hemofilia classifica-se em ligeira, moderada ou grave com base no nível de factor, expresso em percentagem do normal ou unidades internacionais (UI). Habitualmente, os níveis de factor correlacionam-se com a gravidade dos sintomas (Quadro 2). O prognóstico depende fundamentalmente das complicações hemorrágicas e das sequelas resultantes de hemorragias recorrentes.
QUADRO 2 – Classificação das hemofilias A e B
| Grau | Actividade do factor | Tipo de hemorragia | Idade do diagnóstico |
| Ligeiro | ≥ 5 e < 40% (6-39 U/dL) | Frequentemente espontânea | < 1 ano |
| Moderado | ≥ 1 e < 5% (1-5 U/dL) | Relação com trauma mínimo | 1-5 anos |
| Grave | < 1% (< 1U/dL) | Prolongada após trauma grave ou cirurgia | Mais tardia |
Aspectos epidemiológicos
A seguir à doença de von Willebrand, a hemofilia é a coagulopatia hereditária mais frequente, com uma incidência estimada de cerca de 1/10.000 nascimentos. Tratando-se de uma doença recessiva ligada ao cromossoma X, a hemofilia grave afecta quase exclusivamente o género masculino, existindo geralmente antecedentes familiares na linhagem materna. Em cerca de 30% dos casos não existe história familiar (elevada frequência de mutações de novo).
As mulheres portadoras apresentam níveis de factor e manifestações clínicas variáveis. Em casos raros (heterozigotia composta, fenómenos de Lyon, ou perda do cromossoma X) as mesmas podem ter um fenótipo mais grave.
A hemofilia A, mais frequente que a hemofilia B (respectivamente 1/5.000 versus 1/30.000), representa 80-85% do total dos doentes. Está também associada, habitualmente, a maior gravidade
Manifestações clínicas
Embora a hemorragia das mucosas e as equimoses espontâneas/fáceis possam ser os sinais mais precoces da doença, a hemorragia muscular e a hemartrose constituem a manifestação clínica clássica.
Hemofilia grave
Apesar de ser possível o diagnóstico pré-natal (por amniocentese ou punção das vilosidades coriais), na maioria dos casos a hemofilia grave é diagnosticada ao nascer (manifestações hemorrágicas no pós-parto imediato) ou nas primeiras semanas de vida.
Embora a maioria dos doentes não tenha hemorragia significativa no período perinatal, alguns poderão apresentar hematomas extensos ou hemorragia prolongada ao nível da zona cruenta da pele abdominal ao destacar-se o cordão umbilical. A hemorragia intracraniana é rara (3-4% dos casos).
Durante os primeiros meses de vida as manifestações são raras; no entanto, poderá ocorrer hemorragia/hematoma excessivos em situações como erupção dentária, incisão do freio lingual, circuncisão ou aplicação de vacinas por injecção.
Após a aquisição da marcha, à medida que a actividade física aumenta, os hematomas e hemartroses (espontâneos ou após lesões traumáticas minor) tornam-se frequentes, constituindo a principal causa de morbilidade. Com efeito, na criança não tratada, a evolução natural da doença provoca artropatia crónica e incapacitante, resultante de hemartroses recorrentes.
Os hematomas musculares constituem a segunda forma de manifestação mais frequente, podendo estar associados a complicações graves como a síndroma compartimental; por outro lado, as hemorragias localizadas nos grandes músculos (músculos da coxa ou o psoas-ilíaco) podem implicar perdas sanguíneas significativas. Salienta-se que a hemorragia do músculo psoas pode simular sintomatologia compatível com “abdómen agudo”.
As hemorragias do sistema nervoso central, embora raras, constituem a principal causa de morte. Podem ser espontâneas ou pós-traumáticas e, em geral, manifestam-se por cefaleias, vómitos ou alterações do estado de consciência.
Também as hemorragias da via aérea, nomeadamente os hematomas da base da língua e retrofaríngeos, constituem uma emergência pelo risco de obstrução da via aérea. (Figuras 1 a 4)
Embora geralmente de menor gravidade, não é incomum o aparecimento de hematúria, epistaxe ou hemorragia gastrintestinal.
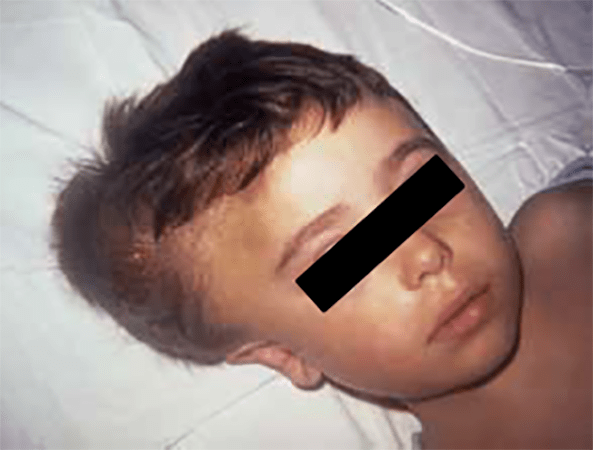
FIGURA 1 – A – Hematoma do couro cabeludo. (NIHDE)
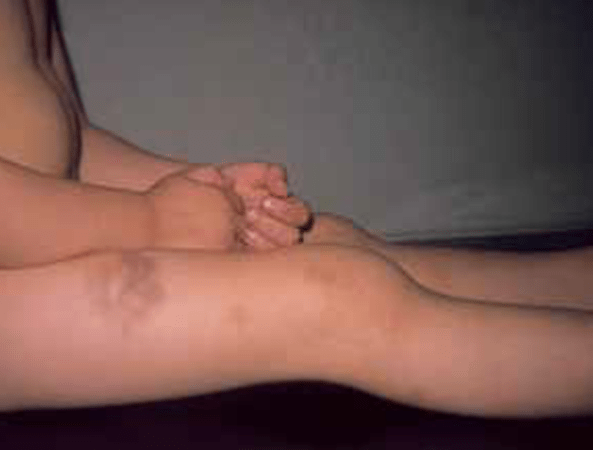
FIGURA 2 – A – Hemartrose do joelho. (NIHDE)
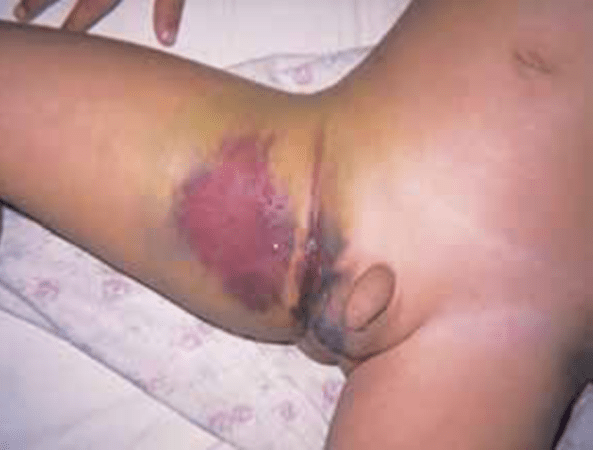
FIGURA 3 – A – Hematoma inguinocrural. (NIHDE)
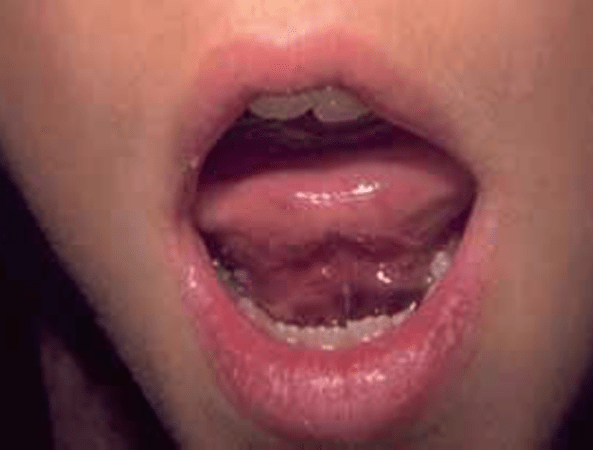
FIGURA 4 – A – Hematoma da base da língua. (NIHDE)
Hemofilia moderada
Nesta forma, as hemorragias espontâneas são raras. Contudo, podem existir hemorragias prolongadas e em desproporção com lesões traumáticas ligeiras.
Hemofilia ligeira
Os indivíduos com hemofilia ligeira apresentam-se, por norma, mais tardiamente (muitas vezes na idade adulta) após, por exemplo, intervenções cirúrgicas, fracturas, feridas contusas de origem traumática, extracções dentárias, ou aplicação de injecções).
Exames complementares
Reportando-nos ao Quadro 1, importa referir que a hemofilia se caracteriza por um prolongamento do tempo de tromboplastina parcial activada (TTPa). O número de plaquetas e o tempo de protrombina (TP) são normais.
Na suspeita de hemofilia, deve proceder-se ao doseamento sérico dos factores de coagulação VIII e IX. Com efeito, o doseamento dos F VIII e F IX permite confirmar o diagnóstico e classificar a hemofilia de acordo com o tipo e gravidade.
Perante um nível baixo de F VIII, é fundamental a documentação de níveis normais do factor de von Willebrand (F vW) para excluir a possibilidade de doença de von Willebrand do tipo 3 (que corresponde a deficiência completa de F vW), sobretudo se existir história de consanguinidade ou ausência de antecedentes familiares de hemofilia.
A distinção entre a doença de von Willebrand do tipo 2N e hemofilia A ligeira implica o estudo da capacidade de ligação do F vW ao F VIII/estudo genético. (ver capítulo seguinte)
O estudo genético está indicado em todos os casos suspeitos de hemofilia. Permite confirmação diagnóstica, estudo familiar e aconselhamento genético, para além de ter valor preditivo quanto ao fenótipo e ao risco de desenvolvimento de inibidores. Após detecção de uma mutação no caso índex, está indicado o estudo genético da família.
Actuação prática
Aspectos gerais
O seguimento destes doentes, por ser complexo, deve ser feito em centros especializados por uma equipa multidisciplinar. A abordagem destes doentes implica cuidados preventivos, terapêutica de substituição com concentrado de factor e tratamento das complicações relacionadas com a doença e com o tratamento.
Administração de concentrados de factor
Nas últimas décadas, após a introdução da terapêutica de substituição com concentrados de factor VIII e IX, a esperança média de vida destes doentes aumentou substancialmente, com diminuição significativa da morbilidade.
Os concentrados de factor constituem a base do tratamento da hemofilia. São fundamentais na profilaxia dos doentes com hemofilia grave e no tratamento da hemorragia aguda. Actualmente estão disponíveis dois tipos de concentrados de factor: os derivados do plasma e os recombinantes (preferencialmente utilizados), estando o uso de plasma fresco ou crioprecipitado contraindicado.
Actuação na hemorragia aguda
Perante uma hemorragia aguda, o principal objectivo é a reposição do factor deficitário para se atingir um nível sérico hemostático. A abordagem terapêutica e dose de factor a administrar dependem do tipo e gravidade da hemorragia.
Na prática, o número de unidades de factor a administrar depende do peso do doente, volume de distribuição do factor e da dose alvo pretendida. O Quadro 3 resume algumas noções importantes a ter em conta na abordagem da hemorragia aguda.
QUADRO 3 – Cálculo para administração de factor em situações de hemorragia aguda
| FACTOR VIII |
| Semi-vida: 8-12 horas Concentrado de factor derivado do plasma ou recombinante: 1 U/kg eleva o nível de factor em 2% Cálculo da dose de F VIII (UI) a administrar = peso do doente (kg) x % de incremento desejado x 0,5 |
| FACTOR IX |
| Semi-vida: 18-24 horas Concentrado de factor derivado do plasma: 1 U/kg eleva o nível de factor em 1% Cálculo da dose de F IX (UI) a administrar = peso do doente (kg) x % de incremento desejado Concentrado de factor recombinante: 1 U/kg eleva o nível de factor em 0,8% (>15 anos) e 0,7% (<15 anos) Cálculo da dose de rF IX (UI) a administrar = peso do doente (kg) x % de incremento desejado x 1,2 (> 15 anos) ou x 1,5 (< 15 anos) |
Na presença de hemorragia confirmada ou suspeita, é fundamental a rápida avaliação e administração de factor como primeira medida (Factor first), não devendo a realização de exames complementares de diagnóstico atrasar a sua administração.
O Quadro 4 descreve mais detalhadamente os níveis de factor desejados e a duração da terapêutica em função da hemorragia específica.
QUADRO 4 – Nível de factor alvo e duração da terapêutica de acordo com o tipo de hemorragia
HEMOFILIA A | HEMOFILIA B | ||
| Tipo de hemorragia | Nível desejado (U/dL) | Nível desejado (U/dL) | Duração (dias) |
| Articular | 40-60 | 40-60 | 1-2 |
| Muscular | 40-60 | 40-60 | 2-3 |
| Ilio-psoas, muscular com compromisso neurovascular ou hemorragia significativa | |||
| · Inicial · Manutenção | 80-100 30-60 | 60-80 30-60 | 1-2 3-5 |
| Laceração profunda | 50 | 40 | 5-7 |
| SNC/Traumatismo Cranioencefálico | |||
| · Inicial · Manutenção | 80-100 50 | 60-80 30 | 1-7 8-21 |
| Orofaringe/pescoço | |||
| · Inicial · Manutenção | 80-100 50 | 60-80 30 | 1-7 8-14 |
| Gastrintestinal | |||
| · Inicial · Manutenção | 80-100 50 | 60-80 30 | 7-14 |
| Renal | 50 | 40 | 3-5 |
| Cirurgia major | |||
| · Pré-operatório · Pós-operatório | 80-100 60-80 40-60 30-50 | 60-80 40-60 30-50 20-40 | 1-3 4-6 6-14 |
| Cirurgia minor | |||
| · Pré-operatório · Pós-operatório | 50-80 30-80 | 50-80 30-80 | 1-5, consoante procedimento |
Outras medidas terapêuticas
Para além da reposição de factor, existem outras medidas terapêuticas que poderão estar indicadas:
1. Acetato de desmopressina (DDAVP)
Este análogo da vasopressina promove a libertação das reservas endoteliais de FVIII, aumentando o nível deste factor em circulação. Os doentes com hemofilia A ligeira e manifestações ligeiras a moderadas podem, muitas vezes, ser tratados apenas com DDAVP, desde que se tenha previamente provado boa resposta.
O DDAVP pode ser administrado por via endovenosa (0,3 mcg/kg/dose, máximo 20 mcg) ou nasal (150 mcg/dose em doente com menos de 50 kg, 300 mcg/dose a partir dos 50 kg). A formulação intranasal indicada tem uma concentração de 1,5 mg/ml, não se encontrando disponível nas farmácias da comunidade (as formulações destinadas à terapêutica da enurese e diabetes insípida têm uma baixa concentração e não devem ser usadas na hemofilia).
Deve ser assegurada a restrição hídrica e não devem ser administradas doses com intervalos inferiores a 24 horas, ou mais de três doses, dada a redução do efeito (taquifilaxia) e o aumento dos efeitos adversos associados (hiponatrémia).
2. Antifibrinolíticos (ácido aminocapróico e ácido tranexâmico)
Estes fármacos inibem a activação do plasminogénio, permitindo a estabilização do coágulo. São úteis nas hemorragias ligeiras das mucosas (oral, gastrintestinal, nasal e ginecológica), podendo dispensar a administração de factor.
Estão contraindicados em hemorragias do tracto urinário pelo risco de formação de coágulos e obstrução vesicouretral. Ambos os fármacos podem ser administrados por via oral, endovenosa ou local (dose de ácido aminocapróico: 75-100 mg/kg/dose; ácido tranexâmico 25 mg/kg/dose, administrados de 6 em 6 horas).
3. Analgesia
Para além das medidas farmacológicas específicas, deve ser instituída precocemente analgesia associada a outras medidas conhecidas pelo acrónimo, do inglês – RICE- (Rest; Ice; Compression; Elevation).
Actuação nas hemorragias com localizações específicas
Hemartrose
Os primeiros sintomas podem ser apenas parestesias ou desconforto local – aura; a reposição precoce de factor previne a lesão tecidual e perpetuação do ciclo hemorragia” lesão” hemorragia.
Hematoma intramuscular profundo
Esta situação exige um elevado nível de suspeição. A hemorragia do psoas-ilíaco pode manifestar-se por dor abdominal, lombar ou referida à articulação coxo-femoral com limitação da extensão do membro (movimentos de rotação mantidos), parestesias da coxa ou outros sinais de compressão do nervo femoral. Os exames de imagem não estão indicados por rotina nas hemartroses e hematomas musculares, nem nas formas aparentemente menos graves; portanto, há que ponderar caso a caso.
Nas hemorragias graves do músculo psoas devem ser realizados exames de imagem após a primeira administração de factor (Factor first). (ver atrás – Actuação na hemorragia aguda)
Hemorragia do sistema nervoso central (SNC)
Constitui uma emergência médica. Os traumatismos cranianos e cefaleias intensas devem ser tratados como hemorragia do SNC.
Nas hemorragias graves do SNC devem ser realizados exames de imagem após a primeira administração de factor (Factor first).
Hematúria
Nesta situação os agentes antifibrinolíticos estão contraindicados. A hematúria microscópica e indolor deve ser tratada com repouso e hidratação vigorosa durante 48 horas. Uma hematúria macroscópica persistente ou hematúria associada a dor ou trauma implicam terapêutica de substituição com concentrados de factor.
Terapêutica em investigação
No início da década de 1990 surgiu a terapêutica génica como uma opção de cura para esta doença monogénica. Contudo, os resultados da investigação não tiveram o êxito esperado até à actualidade.
Por outro lado, os centros de investigação neste campo procuram novas moléculas que permitam prolongar a vida média dos factores que actualmente já são utilizados.
Profilaxia
A profilaxia, indicada na hemofilia grave consiste na administração programada e regular do factor em deficiência. O objectivo é manter um nível mínimo de factor VIII ou IX (entre 1 e 5% ou, genericamente, > 1%) no sentido de diminuir o risco de hemorragia espontânea e de consequente lesão osteoarticular. De facto, os indivíduos com hemofilia moderada raramente contraem artropatia crónica.
São considerados dois tipos de profilaxia em função do momento em que se inicia a respectiva administração:
Profilaxia primária
É iniciada entre o 1 ano e os 2 anos de idade (antes de a criança começar a sofrer de hemartrose ou após a primeira hemorragia articular). Tal medida possibilita uma redução drástica do número de episódios hemorrágicos, preservando as articulações; os inconvenientes são a necessidade de via central de acesso venoso permanente e o elevado custo do factor.
Profilaxia secundária
Considera-se esta modalidade se o tratamento regular contínuo tiver sido iniciado após os 2 anos de idade, ou após duas ou mais hemorragias numa articulação-alvo. De salientar, no entanto, que o esquema profiláctico deve ser individualizado tendo em conta a frequência dos episódios hemorrágicos, a adesão da família e a disponibilidade de acessos venosos.
Complicações
A artropatia hemofílica constituiu uma lesão articular crónica e incapacitante, resultante de hemartroses recorrentes, mais frequentemente numa articulação alvo. Actualmente, a profilaxia primária e o adequado tratamento da hemorragia aguda tornaram esta entidade menos frequente.
A formação de inibidores é uma das complicações mais preocupantes e frequentes no doente com hemofilia. Os inibidores são anticorpos (IgG) contra o factor da coagulação exógeno, tornando a sua reposição ineficaz. Ocorrem aproximadamente em 25% dos doentes com hemofilia A grave, e apenas em 3-5% dos que sofrem de hemofilia B grave, sendo muito menos frequentes na doença moderada ou ligeira. Na hemofilia B poderão também surgir reacções anafilactóides ao factor IX exógeno.
No tratamento dos doentes com baixos títulos de inibidores, as hemorragias podem ser tratadas aumentando as doses de factor VIII ou IX. No entanto, em doentes com elevados títulos, o tratamento das hemorragias implica a utilização dos chamados agentes de bypass (capazes de gerar trombina na ausência de F VIII ou F IX), como o factor VII recombinante activado ou o concentrado de complexo protrombínico activado. Estes doentes são geralmente submetidos a indução de imunotolerância.
As complicações infecciosas relacionadas com derivados do plasma têm fundamentalmente um significado histórico. É importante, no entanto, manter uma vigilância rigorosa dada a possibilidade de transmissão de agentes ainda desconhecidos.
Aconselhamento genético e educação para a saúde
O aconselhamento genético e psicossocial, assim como informação acerca da doença, são fundamentais. A prática de exercício físico deve ser incentivada, procurando evitar desportos de contacto (por exemplo, artes marciais). O doente deve ser alertado para a importância de uma higiene dentária cuidadosa com avaliação periódica pelo estomatologista.
Não existe contraindicação para realização de vacinas, devendo ser cumprido o Programa Nacional de Vacinação (PNV), e estando adicionalmente recomendada a vacina anti-hepatite A. Devem ser cumpridas as recomendações do PNV para indivíduos com alterações da coagulação.
BIBLIOGRAFIA
Croteau SE. Evolving complexity in hemophilia management. Pediatr Clin North Am 2018; 65: 407- 426
Giangrande PLF. Management of haemophilia. Paediatr and Child Health 2015; 25: 350-353
Hoffman R, Benz EJ, Silberstein LE, et al (eds). Hematology: Basic Principles and Practice. Philadelphia: Elsevier, 2018
Kliegman RM, Stanton BF, StGeme JW, Schor NF (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2015
Lanzkowsky P. Manual of Pediatric Hematology and Oncology. Philadelphia: Elsevier Saunders, 2011
Moro M, Málaga S, Madero L. Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood 2015; 125: 2038-2044
Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA (eds). Rudolph´s Pediatrics. New York: McGraw – Hill Medical, 2011
Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia 2013; 19: e1- e47
Stefan DC, Rodriguez-Galindo C (eds). Pediatric Hematology-Oncology in Countries with Limited Resources. Berlin: Springer, 2014
Zimmerman B, Valentino L A. Hemophilia: in review. Pediatr Rev 2013; 34: 289 – 294