Definição e importância do problema
A falência ou insuficiência medular caracteriza-se por disfunção dos precursores hematopoiéticos envolvendo uma ou mais linhagens celulares (eritróide, mielóide e/ou megacariocítica, poupando habitualmente a série linfocitária).
As síndromas de falência medular (SFM), na maioria adquiridas, podem também ser hereditárias; estas últimas, de expressão em idade variável e não necessariamente congénitas ou de manifestação no recém-nascido, correspondem a cerca de 20% dos casos.
A incidência desta patologia é estimada em cerca de 2-6 casos por milhão de habitantes, 2-3 vezes superior na Ásia, sem predomínio de género. Verifica-se uma distribuição bimodal com um pico entre os 10 e os 25 anos, e outro depois dos 60 anos.
Embora se trate de situações clínicas potencialmente fatais, com os avanços das técnicas e acessibilidade de transplante de células progenitoras hematopoiéticas (TCPH), o tratamento das comorbilidades e a disponibilidade de novos fármacos, actualmente o prognóstico é cada vez mais favorável.
Etiopatogénese
A aplasia medular adquirida (AA) deve ser distinguida das formas de falência medular hereditárias (síndromas de falência medular hereditárias – SFMH) e das formas hipoplásicas associadas a síndromas mielodisplásicas (SMD).
As SFMH compreendem cerca de 25-30% dos casos na infância. Distinguir entre AA e as formas hereditárias poderá ser difícil na ausência das manifestações clássicas destas síndromas e/ou história familiar sugestiva. Por outro lado, a distinção entre SMD hipoplásicas e AA pode também constituir um desafio diagnóstico.
Nas formas adquiridas, para explicar a falência (ausência ou défice de produção) da medula óssea não se encontram causas na maioria dos casos (70-80%); este grupo constitui, por isso, as chamadas formas idiopáticas. Nos restantes 20-30% os factores etiológicos encontrados são: exposição a certos fármacos, químicos e vírus infectando os precursores medulares (sobretudo, vírus da imunodeficiência humana/VIH, vírus de Epstein-Barr/VEB, citomegalovírus/CMV, parvovírus B19 e das hepatites).
Os fármacos mais frequentemente associados a insuficiência medular são: cloranfenicol (sistémico), citosina-arabinósido, vincristina, ciclofosfamida, carbamazepina, difenil-hidantoína, indometacina, fenilbutazona, cloroquina, quinidina, acetazolamida, penicilamina, alopurinol, sulfametoxazol-trimetoprim, lítio, metildopa, etc..
Relativamente aos agentes químicos citam-se alguns insecticidas, certos metais de ouro e bismuto, perclorato de potássio, etc..
Na base da anomalia verificada parece estar uma perturbação da imunomodulação por intermédio dos referidos agentes exógenos os quais, activando o sistema imune, conduzem a destruição das células progenitoras/estaminais da medula óssea, sendo esta última substituída por tecido adiposo. Durante anos postulou-se que a patogénese da AA seria imunomediada, dada a resposta à terapêutica imunossupressora, bem como à evidência in vitro de que os linfócitos da medula óssea dos doentes com AA suprimem os linfócitos de medulas saudáveis.
Foram demonstradas as seguintes alterações: aumento de citocinas, e diminuição dos linfócitos T CD4 reguladores e CD8 citotóxicos. Os doentes com AA adquirida têm um número reduzido de linfócitos T reguladores (CD4+/ CD25+). O número destes linfócitos correlaciona-se negativamente com a gravidade da doença, e positivamente com falência do tratamento.
As formas hereditárias (SFMH) fazem geralmente parte de quadros sindromáticos caracterizados pela presença de citopénias e alterações fenotípicas variáveis. As principais, descritas com mais pormenor adiante, são: anemia de Fanconi (AF), anemia de Diamond-Blackfan, síndroma de Schwachman-Diamond (SSD), disqueratose congénita e trombocitopénia amegacariocítica.
Em determinadas situações existe associação a anomalias congénitas (baixa estatura, defeitos no rádio e polegar); contudo, a presença de dismorfismos não é obrigatória.
Está descrito um risco aumentado de doenças malignas (especialmente SMD, leucemia mielóide aguda, tumores sólidos, carcinomas de células escamosas atingindo o pescoço, cabeça e tracto genital).
Manifestações clínicas e exames complementares
A maioria das crianças apresenta-se com sinais e sintomas relacionados com as citopénias (palidez/anemia, infecção/neutropénia, diátese/trombocitopénia), enquanto uma pequena parte é identificada no âmbito de avaliação analítica ocasional (salientando-se a presença de macrocitose como sinal importante de disfunção medular).
As manifestações são explicáveis pelas diferenças de vida média entre plaquetas e leucócitos (mais curta), em relação à dos eritrócitos (mais longa). Assim, surgem primeiramente manifestações de diátese por trombocitopénia (petéquias, equimoses, epistaxes e gengivorragias).
Os sinais de infecção não surgem, em geral, como manifestação inicial excepto nos casos de número de granulócitos < 200/μL; podem estar presentes febre, infecções bacterianas e gengivoestomatite.
A anemia, de instalação lenta (macrocítica normocrómica), traduz-se por palidez da pele e mucosas, astenia, dispneia, entre outros sinais e sintomas. As manifestações clínicas variam em função do grau de pancitopénia. Tipicamente não existem adenopatias nem hepatosplenomegália.
O grau de disfunção (e a gravidade), definido (a) pela celularidade medular e pelo resultado das contagens periféricas de reticulócitos, plaquetas e neutrófilos, pode classificar-se como: moderado/não grave, grave e muito grave. (Quadro 1)
QUADRO 1 – Classificação da falência medular de acordo com a gravidade
Moderada | Celularidade medular < 50%, 2 ou 3 linhagens celulares reduzidas > 6 semanas: |
Grave | Celularidade medular < 25% associada a pelo menos 2 dos seguintes critérios: |
Muito grave | Critérios da AA grave + Neutrófilos < 200/μL |
Os resultados laboratoriais evidenciam sinais sugestivos de insuficiência medular: diminuição do número de reticulócitos (anemia arregenerativa), formas anormais de leucócitos ou elementos mielóides muito imaturos (mais imaturos que bastonetes), plaquetas pequenas, e volume globular médio elevado em desproporção com o valor baixo de reticulócitos. A hemoglobina fetal (HbF) está muitas vezes aumentada.
Classicamente o mielograma evidencia medula óssea hipocelular, rica em gordura, células plasmáticas e reticulares. A biópsia óssea, fundamental para estabelecer o diagnóstico definitivo, permite avaliar o grau de celularidade.
Seguidamente são abordadas de modo sucinto algumas SFMH, bem como outras entidades clínicas a considerar no diagnóstico diferencial.
Anemia de Fanconi (AF)
Trata-se da SFMH mais frequente. De transmissão autossómica recessiva (raramente ligado ao X), decorre de um defeito do mecanismo de reparação do ADN com consequente predisposição a doença maligna (designadamente mielodisplasia, leucemia mieloide e tumores dos tecidos epiteliais). O diagnóstico é feito em geral, entre os 8-10 anos, variando do nascimento aos 30 anos.
Em cerca de 60% dos casos existem anomalias congénitas associadas. Os achados mais típicos e frequentes são: baixa estatura, defeitos dos polegares (por ex. agenésia, dedo supranumerário ou trifalângico – Figura 1), máculas hipopigmentadas ou tipo “café com leite”, anomalias urogenitais (por ex. rim em ferradura), dismorfismo facial (hipoplasia facial, micrognatismo e base nasal alargada).
A disfunção hematológica inicia-se frequentemente com macrocitose, trombocitopénia e evolução para pancitopénia. Estas células têm uma sensibilidade característica predispondo a quebras cromossómicas induzidas por agentes que fazem cross links no ADN, bem como a uma estagnação do ciclo celular na fase G2/M.
O diagnóstico baseia-se:
- Na demonstração, por estudo citogenético, de quebras cromossómicas espontâneas e induzidas por agentes que provocam lesão do ADN (por ex. mitomicina ou dietil-epoxi-butano – DEB); e
- No estudo genético (sequenciação do gene FANC – fundamental em termos de orientação e prognóstico).
Têm indicação para pesquisa de anemia de Fanconi todas as crianças com diagnóstico de aplasia medular idiopática, citopénias de causa desconhecida (sobretudo na presença de macrocitose), mielodisplasia, e em presença de alterações fenotípicas (associação VACTERL, alterações dos membros superiores e/ou genitourinárias e irmãos de crianças com este diagnóstico).
Perante quadro de insuficiência medular grave, as opções terapêuticas são o TCPH ou a utilização de androgénios. De salientar que estas terapêuticas apenas tratam a disfunção medular, razão pela qual a vigilância doutras neoplasias deve ser mantida indefinidamente.
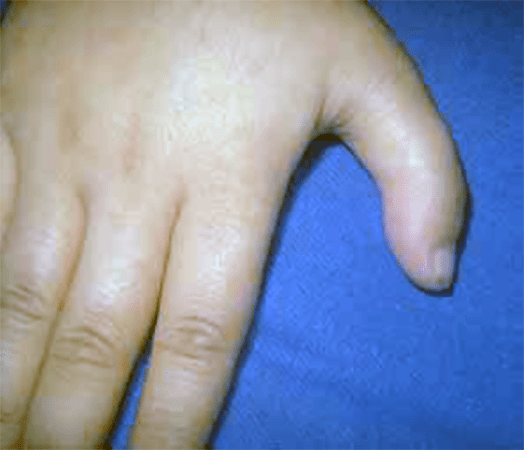
FIGURA 1 – Anemia de Fanconi. Defeito do polegar (com 3 falanges). (NIHDE)
Disqueratose congénita (DC)
A DC é uma síndroma de insuficiência medular congénita, mais frequentemente com um padrão de hereditariedade ligado ao cromossoma X. Na forma clássica, apresenta-se com:
- A tríade clássica de displasia ectodérmica (coloração anormal da pele do pescoço e tronco surgindo habitualmente entre os 6 e os 8 anos), displasia ungueal e leucoplasia da mucosa oral;
- Medula óssea hipocelular afectando as três séries hematopoiéticas.
Trata-se, pois, de uma forma de displasia ectodérmica geneticamente heterogénea, relacionada com defeitos na manutenção dos telómeros (encurtamento progressivo).
As alterações hematológicas (na generalidade, aplasia medular e pancitopénia) surgem em cerca de 50% dos casos por volta dos 10 anos de idade.
Nas formas atípicas as manifestações mucocutâneas surgem após a insuficiência medular.
Por vezes, as manifestações hematológicas, precedendo a pancitopénia, consistem em trombocitopénia ou anemia macrocítica e níveis elevados de hemogobina F.
As alterações genéticas da DC podem ser mutações no gene DKC1, o qual codifica a disquerina, uma proteína implicada na via das telomerases. Outros genes implicados são o TERC e o TERT.
Existem duas variantes do fenótipo mais grave:
- Síndroma de Hoyeraal-Hreidarsson (hipoplasia cerebelosa e atraso do desenvolvimento psicomotor); e
- Síndroma Revesz (semelhante à Hoyeraal-Hreidarsson), mas associada a retinopatia exsudativa.
O diagnóstico de DC implica a presença de 2 dos sinais da tríade clássica associada a uma mutação conhecida ou telómeros curtos. A existência de telómeros curtos no sangue periférico é o achado mais característico, ao ponto de a verificação de telómeros de comprimento normal poder excluir DC.
O diagnóstico de DC pode ser difícil, dado que 50% dos doentes não têm mutação identificada; assim, a medição dos telómeros linfocitários constitui um elemento fundamental para o diagnóstico.
Existe susceptibilidade aumentada para o desenvolvimento de neoplasias como SMD/leucemia mielóide, e tumores sólidos (epiteliais da cabeça e pescoço e região anogenital).
O tratamento é essencialmente de suporte. Poderão ser ponderados o TCPH e, eventualmente, a utilização de androgénios.
Anemia de Diamond-Blackfan
Trata-se duma aplasia eritróide “pura”. A transmissão hereditária é variável; na maioria, trata-se de casos esporádicos.
Segundo os resultados laboratoriais, verifica-se anemia macrocítica tipicamente isolada (podendo, no entanto, coexistir neutropénia e/ou trombocitopénia ligeiras), acompanhada de elevação da hemoglobina fetal (Hb F) e aumento da adenosina deaminase eritrocitária (ADAe).
O aumento da ADAe, de causa não totalmente esclarecida, foi proposto como marcador diagnóstico, com uma sensibilidade de 84% e especificidade de 95%.
A mutação mais frequentemente encontrada é a da proteína ribossomal RPS19 (25%). Em 50% dos casos não existe nenhuma mutação conhecida.
As manifestações surgem no recém-nascido ou durante o primeiro ano de vida em 90% dos casos. Do fenótipo podem fazer parte: baixa estatura/comprimento, polegar trifalângico, pterigium colli, lábio leporino, etc..
A terapêutica passa essencialmente pelo suporte transfusional (sobretudo no primeiro ano de vida) e corticoterapia (60% dos doentes têm resposta, devendo utilizar-se a dose mínima eficaz). Nos casos em que exista elevada dependência transfusional, deverá ser discutida a possibilidade de TCPH.
Existe risco elevado de neoplasias, nomeadamente de mielodisplasia/leucemia mielóide aguda, neoplasia do cólon e de osteossarcoma.
Síndroma de Shwachman-Diamond (SSD)
A etiopatogénese prende-se com uma mutação no gene SBDS, que codifica uma proteína necessária ao metabolismo do ARN.
A SSD é caracterizada por neutropénia (pode estar também presente anemia e trombocitopénia de menor gravidade), insuficiência pancreática exócrina e disostose metafisária.
Em tais situações pode também verificar-se baixa estatura, má progressão ponderal, exantema, alterações dos dentes, bem como sindactilia. Além da história clínica, o doseamento da elastase fecal, do tripsinogénio e da isoamilase pode ser útil para o diagnóstico.
Trombocitopénia amegacariocítica
(ver capítulo sobre Trombocitopénia)
Esta afecção deve ser suspeitada em doentes com trombocitopénia isolada sem os estigmas clássicos da anemia de Fanconi ou da síndroma de trombocitopénia com ausência de rádio (síndroma TAR). Por vezes, são verificadas comorbilidades como microcefalia, baixo peso de nascimento, alterações do neurodesenvolvimento, cardiopatia, anomalias estruturais do sistema nervoso central e anomalias ósseas.
A apresentação clínica na maioria dos casos integra a presença de trombocitopénia isolada, ausência de megacariócitos na medula e evolução para pancitopénia. A hereditariedade pode ser autossómica recessiva ou ligada ao cromossoma X. Em geral, relaciona-se com mutações no gene do receptor da trombopoietina (MPL); contudo, outros genes foram implicados como RUNX1, ANKRD26, MYH9 e PTPN1.
Existe risco de evolução para SMD/LMA. A realização de TCPH, que é curativa, idealmente deve ser realizada previamente à falência medular.
Linfo-histiocitose hemofagocitária
Esta síndroma (hemofagocitária) consiste num quadro de hiperinflamação com desregulação da resposta imune e, consequentemente, inefectiva. Pode ser de causa genética ou secundária a infecções (designadamente por VEB, CMV), a doenças malignas, situações autoinflamatórias ou a doenças metabólicas.
Verifica-se hipercrescimento de histiócitos com consequente fagocitose histiocitária das células sanguíneas nos gânglios linfáticos, medula óssea, fígado e baço.
As manifestações mais típicas são: pancitopénia, hepatosplenomegália, hipertrigliceridémia e pleiocitose no líquido cefalorraquidiano.
Hemoglobinúria paroxística nocturna
(ver Capítulo próprio)
Esta doença, rara na infância, manifesta-se em geral depois dos 5 anos de idade. Caracterizando-se por hemólise intravascular moderada a grave, é mediada pelo complemento e pode estar associada a anemia aplástica, trombose e ferropénia.
Em cerca de 30% dos casos comprova-se hipoplasia medular. A AA pode apresentar-se por expansão de um clone que já perdeu o grupo glicosil fosfatidilinositol (GPI), característico da hemoglobinúria paroxística nocturna (HPN). A citometria de fluxo para pesquisa dos grupos GPI deve ser feita de forma a excluir clones HPN.
Diagnóstico diferencial
O diagnóstico diferencial em geral faz-se com situações que cursam com citopénias associadas a:
- Infiltração ou fibrose medular (leucemia, tumores sólidos como neuroblastoma, doenças de armazenamento, osteopetrose e mielofibrose) acompanhada de pancitopénia.
- Carência de vitamina B12 e ácido fólico em que se verifica destruição intramedular de elementos hematopoiéticos.
- Destruição periférica aumentada (ao nível do baço, fígado ou outros territórios do sistema reticuloendotelial) de células sanguíneas maduras – hiperesplenismo – associada a quadros clínicos diversos tais como hipertensão portal, talassémia, histiocitose, malária e doenças de armazenamento.
São a favor de destruição periférica a presença de reticulocitose, elementos eritróides ou mielóides imaturos no esfregaço de sangue periférico, plaquetas de dimensões aumentadas, elevação do nível de bilirrubina não conjugada e da desidrogenase láctica, e diminuição da haptoglobina.
- Crises aplásticas no contexto de anemia hemolítica crónica (sobretudo relacionada com infecção por Parvovirus B19).
- Citopénia isolada da linhagem eritrocitária – eritroblastopénia transitória da infância na qual ocorre supressão imunológica transitória da eritropoiese, em geral entre os 6 meses e os 3 anos de idade, e de modo insidioso, na criança previamente saudável. A anemia pode ser grave, mas a remissão é espontânea em 1-2 meses. Ao contrário da anemia de Diamond-Blackfan, a percentagem de HbF é normal e a anemia é sempre normocítica.
Tratamento da AA grave e muito grave
As medidas gerais de suporte em situações de insuficiência medular, a ponderar em função do quadro clínico e hematológico, incluem:
- Transfusões de plaquetas (se valor < 10.000/μL em doentes assintomáticos, < 20.000/μL se febre ou se hemorragia activa) e de concentrado eritrocitário (se Hb < 6-7 g/dL ou em doentes sintomáticos).
Os produtos deverão ser irradiados e deverá ser minorado o número de transfusões (com o objectivo de se diminuir o risco de doença do enxerto contra hospedeiro associada à transfusão, e de hemossiderose secundária). Nas adolescentes há que ponderar a supressão da menstruação.
- Antibioticoterapia de largo espectro (por exemplo piperacilina tazobactam em associação, ou não, a aminoglicosídeo, mediante gravidade do caso e de acordo com orientações institucionais) por via parentérica em situações acompanhadas de febre e após colheitas de sangue ou outros produtos para exames culturais.
Na AA o tratamento de primeira linha é o alotransplante medular precoce (com sobrevivência de 85-97%). Na ausência de dador familiar, ou não relacionado histocompatível, deve ser iniciada imunossupressão com globulina antitimócito de cavalo (melhor resposta que a de coelho), associada a ciclosporina A de acordo com as recomendações internacionais. A terapêutica imunossupressora está associada a taxas de resposta entre 60-75%, com uma sobrevivência de 80-90% e recidiva de 10-30%.
Como segunda linha poderão ser utilizados outros imunossupressores, ser considerada a utilização de agonistas rTPO ou outras modalidades de transplante (haploidêntico).
O tratamento da AA moderada é controverso dado que a evolução é variável; certa parcela de casos evolui para AA severa e outra parcela regride espontaneamente.
Nas SFMH, para além da terapêutica específica para a insuficiência medular deve providenciar-se:
- Vigilância clínica rigorosa na perspectiva sobretudo de neoplasias; e
- Aconselhamento genético.
BIBLIOGRAFIA
Alter BP, Giri N, Savage SA, et al. Cancer in dyskeratosis congenita. Blood 2009; 113: 6549–6551
Barone A, Lucarelli A, et al. Diagnosis and management of acquired aplastic anemia in childhood. Guidelines from the Marrow Failure Study Group of the Pediatric Haemato-Oncology Italian Association (AIEOP). Blood Cells, Molecules, and Diseases 2015; 55: 40–47
Chirnomas S, Kupfer G. The inherited bone marrow failure syndromes. Pediatr Clin North Am 2013; 60: 1291–1310.
Freitas O, Braga L, et al. Anemias aplásticas. Revisão de 10 anos (2002-2011) na Unidade de Hematologia do Hospital Dona Estefânia, Lisboa. Reunião clínica de 10 de Abril de 2012
Green AM, Kupher GM. Fanconi anemia. Hematol Oncol Clin North Am 2009; 23: 193-214
Hoffman R, Benz EJ, Abutalib SA (eds). Hematology. Philadelphia: Elsevier, 2018
Kliegman RM, Stanton BF, StGeme JW, Schor NF (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2015
Korthof E, Bekassy A, Hussein A. Management of acquired aplastic anemia in children. Bone Marrow Transplantation 2013; 48: 191–195
Marsh JCW. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol 2009; 147: 43-70
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Nathan DG, Orkin SH, Look AT, Ginsburg D (eds). Nathan and Oski’s Hematology of Infancy and Childhood. Philadelphia: Saunders, 2009
Noronha SA. Aplastic and hypoplastic anemias. Pediatr Rev 2018; 39: 601-611. DOI: 10.1542/pir.2017-0250
Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA (eds). Rudolph’s Pediatrics. New York: McGraw-Hill Medical, 2011
Samarasinghe S, Webb DK. How I manage aplastic anaemia in children. Br J Haematol 2012; 157: 26-40
Savasan S. Acquired aplastic anemia: what have we learned and what is in the horizon? Pediatr Clin North Am 2018; 65: 597-606
Scheinberg P. Aplastic anemia: therapeutic updates in immunosuppression and transplantation. Hematol/Educ Program Am Soc Hematol Am Soc Hematol Educ Program 2012: 292-300
Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Reviews 2010; 240: 101-122
Stefan DC, Rodriguez-Galindo C (eds). Pediatric Hematology-Oncology in Countries with Limited Resources. Berlin: Springer, 2014
Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood 2014;124: 2775-2783
Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood 2010;116: 3715-3723