Introdução
A hemoglobina (Hb) é a proteína transportadora de oxigénio para todos os tecidos do organismo. É formada por quatro cadeias peptídicas (globinas), cada uma delas ligada a um grupo heme. Diferentes estruturas de Hb evoluem ao longo do desenvolvimento fetal, embrionário, e dos primeiros meses de vida até se atingir a estrutura Hb A (2 cadeias α e 2 cadeias β – α2β2), que predomina na idade adulta. (Quadro 1)
No embrião predominam as Hb Gower-1 (ζ2ε2), Gower-2 (α2ε2) e Portland (ζ2γ2). A partir da décima semana de gestação e até ao sexto mês de vida, predomina a Hb fetal (α2γ2). Com a aproximação do nascimento, passa a ser produzida a Hb A e uma pequena quantidade de Hb A2 (α2δ2). Em estados patológicos associados a eritropoiese ineficaz como a talassémia e a drepanocitose, verifica-se uma diminuição na produção de Hb A com aumento da síntese da Hb F (recapitulação da ontogenia). Cada variante de Hb tem propriedades distintas que conferem vantagens específicas durante a respectiva fase de desenvolvimento.
Com efeito, o equilíbrio entre os pares de globinas é fundamental para os processos metabólico e fisiológico do glóbulo vermelho. A falta de produção de uma cadeia de globina determina que não se forme quantidade normal de Hb; por consequência, os eritrócitos são microcíticos e hipocrómicos. Por outro lado, a cadeia de globina não afectada continua a produzir-se em quantidade normal, do que resulta: – um excedente desta, que precipita e causa lise prematura, inclusivamente na medula óssea (eritropoiese ineficaz); ou – formação de tetrâmeros de uma só cadeia (Hb H, com quatro cadeias β, ou Hb de Bart, com quatro cadeias γ) que não podem libertar oxigénio como acontece com a Hb normal e precipitam nas células em qualquer momento do seu desenvolvimento. Os eritrócitos resultantes têm consequentemente vida média encurtada, destruindo-se de forma prematura, sobretudo no baço.
As diferentes cadeias de globina são codificadas por dois grupos génicos: o grupo β localiza-se no cromossoma 11 e contém um gene β, um δ, um ε e um γ; o grupo α localiza-se no cromossoma 16 e contém 2 genes α e 1 ζ. (Quadro 1)
QUADRO 1 – Hemoglobina humana
| Fase da ontogénese | Hemoglobina | Estrutura | % |
| Embrionária | Gower-1 Gower-2 Portland | ζ2ε2 α2ε2 ζ2γ2 | 20-40 10-20 5-20 |
| Fetal | Fetal | α2γ2 | 90-100 |
| Pós-nascimento | A A2 Fetal | α2β2 α2δ2 α2γ2 | 96-98 2-4 0-1 |
As hemoglobinopatias encontram-se entre as doenças autossómicas recessivas mais comuns. Estima-se que mais de 250 milhões de pessoas (cerca de 4,5% da população mundial) sejam portadoras de uma alteração da hemoglobina.
As hemoglobinopatias são classicamente divididas em quantitativas e qualitativas.
As hemoglobinopatias quantitativas ou de síntese, vulgarmente conhecidas como talassémias, resultam da síntese diminuída ou ausente de uma globina estruturalmente normal.
As hemoglobinopatias qualitativas ou de estrutura incluem hemoglobinas com alteração da função ou das suas propriedades físicas ou químicas que resultam da mutação genética, determinando substituição de um aminoácido numa das cadeias polipeptídicas. As hemoglobinas anormais comuns – S, C, D, E provêm de substituições de um aminoácido nas cadeias β; são exemplos as diferentes formas de doença falciforme (Hb S) e outras adiante descritas.
Nalguns países procede-se ao rastreio neonatal das hemoglobinopatias.
ALTERAÇÕES QUANTITATIVAS DA HEMOGLOBINA – TALASSÉMIAS
Definição
As talassémias são anemias hemolíticas congénitas que resultam de anomalia da síntese de uma ou mais subunidades das cadeias de globina que formam a hemoglobina. A cadeia da globina afectada estará presente em quantidades inferiores às normais ou, inclusivamente, ausente.
Patogénese e classificação
Assim, as talassémias classificam-se conforme a cadeia de globina cuja síntese está afectada:
- Alfa talassémia <> síntese de globina α reduzida ou ausente;
- Beta talassémia <> síntese de globina β reduzida ou ausente;
- E assim, sucessivamente, para as restantes globinas (por ex. γ ou outras) que, mesmo se presentes, mas deficitárias, evidenciam estrutura normal.
Assim, designando-se as talassémias pelo nome cadeia de globina ausente ou deficitária, na representação gráfica utilizam-se os seguintes símbolos: por ex. αº ou βº se a síntese estiver ausente; e α+ ou β+ se a síntese estiver reduzida.
A síntese diminuída ou ausência de uma das globinas (α, β ou γ) determina excesso relativo da outra; por outro lado, as referidas globinas não emparelhadas são instáveis, precipitam no interior do glóbulo vermelho e provocam lesões oxidativas da membrana plasmática, resultando em hemólise. A gravidade da hemólise depende da quantidade e da variante de globina mutada.
As cadeias α não emparelhadas são insolúveis, ao contrário das cadeias β e γ, que mantêm alguma capacidade de formar tetrâmeros da mesma globina.
Numa das formas de beta talassémia, chamada major, a ausência de cadeias β cria um grande desequilíbrio entre as globinas, sendo os eritroblastos destruídos ainda na medula óssea, antes de entrarem em circulação, levando a uma anemia grave. Nestes doentes, há um aumento compensatório na actividade eritropoiética medular que, no entanto, nunca é completo – é a chamada eritropoiese ineficaz.
Diagnóstico pré-natal
Na actualidade é possível detectar a mutação responsável por cada tipo de talassémia a partir de amostra de líquido amniótico ou de biópsia coriónica obtida entre as semanas 8 e 18 de gestação. O diagnóstico realiza-se através do estudo do ADN, em geral com técnicas de PCR (reacção em cadeia da polimerase).
Beta talassémia – generalidades
Compreende um grupo de anemias hemolíticas hereditárias causadas por anomalia na síntese da globulina β da Hb, com produção de excesso relativo de cadeias α de globina.
Nesta forma de talassémia, e na sua maioria, verificam-se mutações pontuais que afectam poucas bases, alterando-se a expressão do gene da cadeia β da globina. Foram descritas mais de 100 mutações, mas somente cerca de 5 ou 6 delas afectam mais de 90% dos pacientes; as mutações predominantes variam conforme as etnias.
A beta talassémia é prevalente em populações do Mediterrâneo, Médio Oriente, Centro, Sul e Sueste da Ásia e Sueste da China. Resulta habitualmente de mutações pontuais em várias regiões do gene β, sendo muito raras as deleções.
As manifestações clínicas, muito variáveis, dependem do desequilíbrio entre a síntese das globinas α e β, ou seja, da quantidade de cadeias α não emparelhadas. Mutações na região codificadora do gene β levam à ausência de síntese da globina desse alelo (β0), enquanto mutações noutras regiões do gene levam a redução ligeira ou moderada da respetiva síntese (β+). Sob o ponto de vista clínico, as beta-talassémias classificam-se em major, minor e intermédia.
Beta talassemia major (anemia de Cooley)
Etiopatogénese e clínica – A beta talassémia major resulta de homozigotia ou heterozigotia composta (β0/β0) com défice grave ou ausência de síntese de globina β nos glóbulos vermelhos. Uma vez que as variantes de Hb embrionárias e a Hb F não possuem cadeias β, os sintomas surgem a partir dos 6 meses de idade.
A ausência de síntese das cadeias β da globina determina anemia hemolítica intensa e crónica. Como mecanismo compensador do organismo verifica-se hiperplasia do tecido hematopoiético (expansão das cavidades medulares, hepatosplenomegália) e aumento da absorção digestiva do ferro.
Os sinais iniciais são palidez, icterícia e hepatosplenomegália ligeiras (por eritropoiese extramedular) que são mais notórias pelos 2 anos de idade. Concomitantemente verifica-se compromisso progressivo do crescimento com agravamento da síndroma anémica (palidez “terrosa”) ao longo dos anos e litíase biliar.
Poderão surgir progressivamente alterações cardíacas como resultado do estado hiperdinâmico secundário à anemia de gravidade progressiva, e hemossiderose miocárdica, a principal causa de mortalidade por insuficiência cardíaca. Igualmente, alterações endócrinas (diabetes mellitus, hipotiroidismo, diminuição da actividade da somatomedina) possivelmente em relação com o depósito do ferro e hipóxia crónica.
De salientar que o ferro acumulado provém, tanto da degradação da Hb, como da sua absorção intestinal aumentada; como consequência poderá instalar-se um quadro de deposição visceral generalizada de Fe. (Figura 1)

FIGURA 1 – Etiopatogénese da ß-talassémia. Adaptado de Weatherall DJ, 1998
Por volta dos 4 a 6 anos passa a ser progressivamente notório um conjunto de características dismórficas craniofaciais ou fenótipo sui generis: prognatismo do maxilar superior, retrognatismo do inferior, procidência das bossas frontais, turricefalia.
As alterações esqueléticas são o resultado da hiperplasia da medula óssea que determinam alargamento do espaço medular, com adelgaçamento da cortical e marcada osteoporose. Com efeito, estas alterações são mais evidentes nos ossos do crânio (padrão radiográfico “crânio em escova”, citado a propósito da drepanocitose) (Figura 3) e face dando origem à típica fácies asiática ou “de esquilo” (crescimento excessivo do maxilar, malares e gengivas salientes, depressão da ponte do nariz e protrusão dos dentes anteriores). Estas alterações e a eritropoiese extramedular são o epifenómeno de eritropoiese ineficaz. (Figuras 2 e 3)
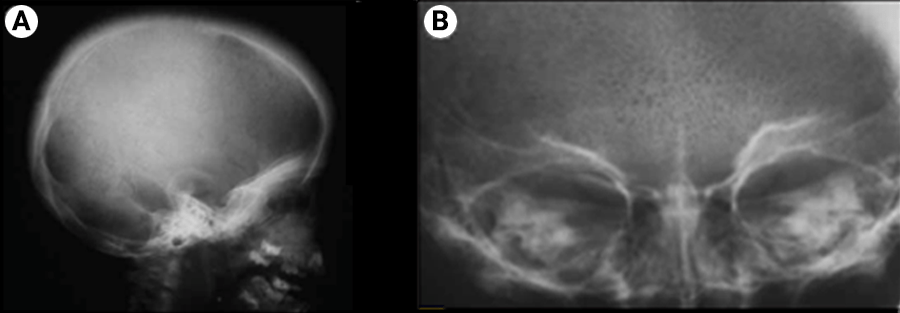
FIGURA 2 – Radiografia do crânio: A – Crânio em escova; B – Sinais de hiperplasia medular. (NIHDE)
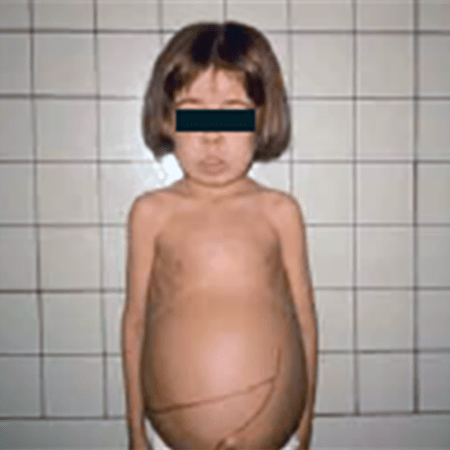
FIGURA 3 – Fácies talassémica. Hepatosplenomegália exuberante*. (NIHDE)
*Este quadro exuberante, observado há três décadas, é hoje raro dados os progressos verificados no diagnóstico e na terapêutica. (Cortesia da Profa MG Gomes da Costa ao NIHDE).
A expansão eritróide nos ossos longos, crânio e ossos faciais leva a adelgaçamento cortical com risco de fracturas ósseas e a alterações craniofaciais características (há um agravamento da hemólise após exposição a situações de estresse oxidativo e susceptibilidade a crises aplásicas por parvovírus B19).
Achados analíticos – Verifica-se anemia grave com microcitose e hipocromia (índice de Mentzer < 11,5) e reticulocitose. O esfregaço de sangue periférico apresenta anisopoiquilocitose, hipocromia, microcitose, células em alvo, eritroblastos e ponteado basófilo. O estudo das hemoglobinas por HPLC (high performance liquid chromatography) revela a ausência de Hb A e predomínio de Hb F.
No recém-nascido os valores iniciais de Hb são normais, diminuindo progressivamente para atingirem progressivamente valores inferiores a 5 g/dL nos primeiros meses de vida. O número de reticulócitos varia entre 2 e 8%.
O exame da medula óssea mostra sinais de hipercelularidade com intensa hiperplasia eritróide e diseritropoiese. Os depósitos de ferro estão muito aumentados. A resistência osmótica está também aumentada. A siderémia está elevada assim como a saturação da transferrina e a ferritina.
A avaliação da cinética do ferro mostra um padrão de eritropoiese ineficaz.
Tratamento – O suporte transfusional regular, em regra mensal, é o principal tratamento dos doentes com beta talassémia major. Permite suprimir a eritropoiese endógena ineficaz e prevenir a restrição do crescimento e as alterações esqueléticas. A fenotipagem eritrocitária alargada antes da primeira transfusão é fundamental para minorar o risco de aloimunização. A esplenectomia está indicada em situações de esplenomegália sintomática ou de necessidades crescentes de suporte transfusional.
Existe risco elevado de tromboembolismo após a esplenectomia, pelo que a profilaxia com antiagregante plaquetar deve ser considerada nalguns casos, sobretudo na presença de trombocitose.
A forma mais simples de quantificar a sobrecarga de ferro baseia-se na contagem do número total de transfusões. Os níveis séricos de ferritina correlacionam-se com os depósitos corporais de ferro e constituem outro método simples, embora aqueles sejam influenciados por estados de infecção/inflamação. A ressonância magnética (RM) é um método não invasivo de quantificar os depósitos corporais de ferro, em particular o ferro cardíaco, que pode ser monitorizado por RM cardíaca T2*: -níveis de T2* inferiores a 20 ms associam-se a um risco aumentado de insuficiência cardíaca.
A quelação do ferro deve ser iniciada após 20-25 transfusões, ferritina > 1000 ng/mL ou evidência imagiológica de sobrecarga de ferro. Existem actualmente três fármacos aprovados em doentes pediátricos: desferroxamina (via subcutânea ou endovenosa), deferiprona (via oral) e deferasirox (via oral). Cada um apresenta um perfil particular de características farmacocinéticas e farmacodinâmicas e de efeitos adversos.
Os indivíduos com sobrecarga de ferro e sob quelação (sobretudo sob desferroxamina) têm um risco elevado de bacteriémia, nomedamente por Yersinia enterocolitica; por isso, a suspensão da quelação é obrigatória durante qualquer intercorrência infecciosa.
O transplante alogénico, actualmente o único tratamento curativo, tem excelentes resultados em indivíduos jovens sem doença avançada.
Beta talassémia intermédia
Etiopatogénese e clínica – Esta forma, correspondendo geralmente a 2 genes da β-globina afectados, integra fundamentalmente os genótipos β+/β+ ou β+/βº. A cromatografia da Hb revela valores aproximados de HbA: 20-40%, Hb A2: 5% e HbF: 60-80%.
Caracteriza-se por um fenótipo clínico heterogéneo em que há anemia hemolítica moderada a grave (com icterícia e esplenomegália moderadas), parcialmente compensada sem necessidade de suporte transfusional regular.
Alguns doentes mantêm-se assintomáticos sem transfusões, e outros necessitam de transfusões periódicas. Verifica-se frequentemente hepatosplenomegália, litíase biliar e estigmas de expansão medular. Importa sublinhar que a eritropoiese ineficaz com aumento da absorção de ferro leva a sobrecarga importante de ferro mesmo na ausência de suporte transfusional, pelo que a quelação deve ser considerada.
Achados analíticos – Observa-se um grau variável de anemia com glóbulos vermelhos hipocrómicos e microcíticos, e células em alvo no esfregaço de sangue periférico. O valor de Hb por vezes atinge 8 g/dL.
Tratamento – Não existem orientações quanto ao início de suporte transfusional nestes doentes; a decisão de transfundir, bem como a sua periodicidade, devem ser individualizadas. Níveis basais de Hb de 7 a 9 g/dL são eficazes na maioria dos doentes; contudo, outros podem necessitar de valores mais elevados, sobretudo durante o crescimento, gravidez ou intercorrências infecciosas.
A utilização de hidroxicarbamida, evidenciando resultados satisfatórios num subgrupo de doentes com aumento dos níveis basais de Hb, tem permitido diminuir a necessidade transfusional.
A esplenectomia melhora a anemia, mas existe, para além do risco infeccioso inerente, risco muito elevado de tromboembolismo; por isso, deverá ser evitada, se possível.
Beta talassémia minor (ou traço talassémico)
Etiopatogénese e clínica – Ocorre em indivíduos heterozigóticos assintomáticos. Verifica-se apenas um alelo mutado. Esta forma a que corresponde ausência de expressão de 1 gene da β-globina afectado (β°) (genótipo ββ0), caracteriza-se por escassez de sinais e sintomas.
Encontra-se distribuída em grupos étnicos da zona mediterrânica (Itália e Grécia), Sueste asiático e em populações de origem africana.
Achados analíticos e diagnóstico diferencial – São comprovados parâmetros de hipocromia e microcitose, sem anemia (ou, existindo, muito ligeira), ou de hemólise. A fracção A2 da Hb está aumentada (habitualmente 4-7%) e em 50% dos casos verifica-se também aumento da HbF.
É importante o diagnóstico diferencial com ferropénia e com talassémia εγδβ. A Hb A2 pode estar falsamente normal na presença de ferropénia grave: nestes casos, a cromatografia deve ser repetida após a reposição das reservas de ferro.
Os raros indivíduos heterozigóticos para a deleção εγδβ apresentam uma anemia hemolítica moderadamente grave durante o período neonatal, a qual melhora durante os primeiros anos de vida. Durante a adolescência são identificadas alterações hematológicas semelhantes às da beta talassemia minor, à excepção da electroforese de hemoglobinas, que evidencia Hb A2 normal.
Actuação prática – Os portadores de β-talassémia minor não necessitam de qualquer tratamento ou vigilância específicos. No entanto, é fundamental alertar e sensibilizar os pais e crianças para a necessidade de rastreio de hemoglobinopatias nos futuros companheiros de forma a reduzir a incidência das formas mais graves da doença.
Não deverá ser administrado ferro sob pena de agravamento da tendência para hemossiderose.
Alfa talassémia
A alfa talassémia, que integra quadros de diagnóstico mais difícil porque não são acompanhados de alterações significativas da HbA2 e Hb F, tem uma elevada prevalência em África, Sueste Asiático e Índia.
Recorda-se que os indivíduos normais possuem quatro genes α activos responsáveis pela síntese de cadeias α (2 em cada cromossoma 16).
Nas α – talassémias, a que corresponde largo espectro de síndromas, há diminuição da síntese de cadeias α levando a anemia, salientando-se que o grau de anemia é directamente proporcional ao número de deleções.
Em função do número de genes activos afectados da globulina α (mutações relacionadas com deleções) são descritas quatro síndromas. (Quadro 2)
Como cada indivíduo possui quatro cópias do gene α, as alterações são mais heterogéneas do que na β talassémia.
O gene α é expresso desde as primeiras semanas de vida, razão pela qual a alfa talassémia se manifesta na vida fetal e pós-natal. As manifestações clínicas dependem do número de alelos funcionantes. (Quadro 2)
QUADRO 2 – Síndromas de alfa talassémia
Síndroma | Número de genes afectados com mutações | Alterações laboratorais | Padrão da hemoglobina |
Portador silencioso | 1 (-α/αα) | Sem anemia | Hb Bart 1-2% |
Traço talassémico | 2 (–/αα; cis) | Anemia ligeira | Hb Bart 5-10% |
Doença Hb H | 3 (–/-α) | Anemia moderada | Hb H 10-30% |
Hidropisia fetal | 4 (–/–) | Anemia grave | Hb Bart 97% |
Cerca de 1 em cada 3 afro-americanos é portador silencioso de alfa talassémia. Tais indivíduos não têm anemia nem hemólise e apresentam parâmetros hematológicos (volume globular médio e hemoglobina globular média) no limite inferior da normalidade.
O traço talassémico pode ocorrer de duas formas (–/αα ou cis, comum na Ásia, ou -α/-α ou trans, comum em África) e associa-se a ligeira anemia hipocrómica e microcítica. O esfregaço de sangue periférico mostra glóbulos vermelhos hipocrómicos e microcíticos e células em alvo. A cromatografia da Hb é normal, pelo que o diagnóstico definitivo só é possível mediante estudo molecular.
À semelhança dos portadores silenciosos, estes indivíduos não necessitam de vigilância ou tratamento específicos. Nos indivíduos asiáticos (–/αα) devem ser feitos estudos familiares e aconselhamento genético de forma a evitar a Hb Barts – hidropisia fetal que é incompatível com a vida e pode acarretar graves complicações para a grávida.
Na doença da Hb H apenas um dos quatro genes está activo (–/-α). Existe, portanto, deleção de três genes.
Na vida adulta predomina a HbA com 5-30% de Hb H; no período neonatal predomina a HbF com 10-20% de Hb Bart, sendo vestigial o teor de Hb H.
O quadro clínico é semelhante ao da talassémia major ou da intermédia.
No esfregaço de sangue periférico, é característica a presença de inclusões citoplasmáticas após coloração com azul cresil. Alguns indivíduos são dependentes de transfusão enquanto outros têm uma anemia mais ligeira. Há o risco de exacerbação após exposição a estresse oxidativo com maior susceptibilidade a aplasia/hipoplasia no decurso de intercorrências infecciosas. Como a eritropoiese ineficaz é pouco significativa, a sobrecarga de ferro ocorre mais lentamente do que a observada na beta-talassémia.
A hidropisia fetal por Hb Bart caracteriza-se pela ausência de cadeias α (por deleção dos quatro genes respectivos, com genótipo –/–) e formação de tetrâmeros de cadeias γ (γ4), com uma hemoglobina com elevada afinidade pelo oxigénio levando a hipóxia celular fetal, hidropisia fetal (hepatosplenomegália e anasarca) com elevado risco de morte fetal e neonatal. Estão descritos alguns casos de sucesso de transfusão intra-uterina seguida de regime transfusional regular e transplante alogénico de células progenitoras hematopoiéticas.
Existem ainda outras síndromas de alfa-talassémia causadas por mutações de novo, ou adquiridas, alterando a expressão dos genes da α-globina:
- A síndroma de alfa-talassémia com atraso mental, integrando duas formas:
- uma, associada a deleções extensas no cromossoma 16 envolvendo os genes da α-globina (ATR16, por mutação de novo);
- outra, com genes da α-globina estruturalmente normais mas com alteração de um factor de transcrição localizado no cromossoma X, fundamental para a regulação da expressão dos respectivos genes (ATRX). Estão presentes dismorfias faciais e/ou genitais a que se associa uma forma moderada de Hb H, geralmente menos grave que a verificada na doença da HbH.
- A doença da Hb H adquirida associada a síndromas mielodisplásicas. Também nesta situação os genes da α-globina são estruturalmente normais. As alterações decorrem de mutações somáticas adquiridas no gene ATRX, com fenótipo mais grave.
ALTERAÇÕES QUALITATIVAS DA HEMOGLOBINA – VARIANTES ESTRUTURAIS DA HEMOGLOBINA
Foi referido anteriormente que as hemoglobinopatias qualitativas ou de estrutura incluem hemoglobinas que resultam da mutação genética, determinando substituição de um aminoácido numa das cadeias polipeptídicas, sendo que nas mais comuns tal substituição ocorre nas cadeias β. Acrescenta-se que o conjunto de genes necessários para a produção de hemoglobina localizam-se no braço curto dos cromossomas 16 e 11.
No Quadro 3 descrevem-se as bases moleculares de algumas variantes de hemoglobina relacionadas com diferentes cadeias polipeptídicas afectadas, para além da cadeia β, em que se verificou substituição de aminoácido. As variantes que não evidenciam repercussão clínica relevante são designadas por alguns autores como hemoglobinoses.
QUADRO 3 – Algumas variantes de hemoglobina
Substituição de um único aminoácido
|
| Deleções de aminoácidos: Freiburg |
| Fusão de hemoglobinas: δβ (Lepore), βδ, γβ |
No Quadro 4 são sistematizadas as variantes de hemoglobina com relevância clínica, a seguir abordadas.
QUADRO 4 – Variantes de hemoglobina com relevância clínica
| Doença de células falciformes/Hb S, e outros defeitos- Hb S, Hb C, Hb E, Hb SC |
| Hemoglobinas instáveis |
| Hemoglobinas com alta ou baixa afinidade para o oxigénio |
| Hemoglobinas M |
| Variantes estruturais com fenótipo simile talassémico α e β |
DOENÇA DE CÉLULAS FALCIFORMES
Definição
Doença de células falciformes é o nome dado a um conjunto de defeitos da Hb em que se verifica a presença da chamada HbS. Trata-se duma hemoglobinopatia qualitativa ou de estrutura resultante de mutação genética, determinando substituição de um aminoácido numa das cadeias polipeptídicas, neste caso por mutação de gene da globina β no cromossoma 11.
Aspectos epidemiológicos e classificação
Sendo a hemoglobinopatia mais frequente (nos EUA ~1/600 recém-nascidos), tal entidade, com diversidade de apresentação clínica, evidencia morbilidade e mortalidade muito significativas, partilhando com as síndromas talassémicas muitas características.
Com efeito, em relação à respectiva distribuição, ambas apresentam uma elevada frequência nos países do Mediterrâneo, Médio Oriente, Índia e África Oriental e Equatorial, nos quais constituem um problema de saúde pública. Afectando fundamentalmente a raça negra, a sua distribuição na Europa e Américas, incluindo Caraíbas, explica-se pelo fluxo migratório desde há mais de cinco séculos, o que tem implicações de ordem genética.
Ambas coincidem com as regiões onde a malária pelo Plasmodium falciparum foi endémica, o que confere uma selecção natural responsável pela manutenção e perpetuação dos genes.
Como apresentado no Quadro 5, a doença de células falciformes inclui diversos genótipos a que corresponde sintomatologia variada (fenótipo), desde a forma homozigótica – Hb SS ao chamado traço falciforme – HbAS, este último assintomático ou com manifestações benignas. Como se depreende pela observação do mesmo quadro, a proporção de Hb F, Hb A1, Hb A2 nas hemoglobinopatias S é variável.
A anemia de células falciformes (ACF), também denominada drepanocitose, de transmissão autossómica recessiva, é a forma mais grave do espectro da doença de células falciformes.
A prevalência da homozigotia (Hb SS), que resulta da mutação dos genes da globina β no cromossoma 11 de ambos os progenitores, varia entre 0,2% nos afro-americanos e 6% em certas regiões de África. A heterozigotia, que resulta da mutação de apenas um gene da globina β de um dos progenitores, afecta cerca de 12% dos afro-americanos e cerca de 40% da população em certas regiões de África. Dados relativos a Portugal são limitados. Estimativas recentes apontam para cerca de 600 indivíduos homozigóticos no nosso país e para um gradiente crescente Norte-Sul na sua prevalência, explicado pelos fenómenos migratórios das populações (imigração de escravos africanos no século XV e imigração de países de elevada prevalência nos últimos anos).
QUADRO 5 – Características das diferentes formas de doença de células falciformes
| Tipo de Hb | Gravidade clínica | Hb S (%) | Hb F (%) | Hb A2 (%) | Hb A (%) | Valor Hb (g/dL) | VGM (fL) |
| Anemia de células falciformes | |||||||
| SS | Normalmente grave | > 90 | < 10 | < 3,5 | 0 | 6-9 | > 80 |
| Heterozigótias duplas | |||||||
Sβ0 Sβ+ SC S– PHHF | Moderada a grave Ligeira a moderada Ligeira a moderada Assintomática | > 80 > 60 50 < 70 | < 20 < 20 < 5 > 30 | > 3,5 > 3,5 0 < 2,5 | 0 10-30 0 0 | 6-9 9-12 10-15 12-14 | < 70 < 75 75-85 < 80 |
| Traço falciforme | |||||||
| AS | Habitualmente assintomática | 30-40 | 1,5 | < 2,5 | 60-70 | 11-12 | 85 |
| β° = gene talassémico com ausência de síntese da cadeia β; β+ = gene talassémico com diminuição de síntese da cadeia β; VGM = volume globular médio; S- PHHF: = Persistência hereditária de Hb fetal (PHHF) Nesta entidade (de que existem descritas > 20 variantes), resultante de deleção ou mutação originando défice de produção de cadeias β ou δ ou de ambas, verifica-se incapacidade para a síntese da cadeia γ na fase de transição da vida intrauterina para a extrauterina; de tal resulta a manutenção durante toda a vida de níveis elevados de Hb F. As manifestações são silenciosas (anemia e microcitose ligeiras). | |||||||
Etiopatogénese
A base molecular da anemia de células falciformes (ACF), a forma mais grave da doença de células falciformes, é a substituição de um único aminoácido na cadeia da β-globina (valina por ácido glutâmico na sexta posição originando a HbS ou α2 βs2); tal acarreta modificação da forma do eritrócito, perdendo a forma bicôncava e adquirindo a forma em foice (falciforme) donde deriva o nome da doença.
A HbS (α2 βs2) tem a propriedade única e própria da variante β6 Glu-Val de se polimerizar quando desoxigenada, processo central da vasoclusão e causa primária de certas manifestações clínicas. Na polimerização poderão interferir factores agravantes (como a hipóxia e acidose, desidratação, elevação da temperatura, factores genéticos, etc.), ou atenuantes como por exemplo a percentagem de hemoglobina fetal (Hb F), a qual constitui o inibidor mais potente da despolimerização da desoxi-hemoglobina.
Os referidos eritrócitos falciformes têm fragilidade excessiva, menor deformabilidade (eritrócitos mais rígidos), vida média muito encurtada (cerca de 20 dias), circulando com dificuldade na microcirculação por hiperviscosidade, aderindo à parede do endotélio e lesando-a (vasculopatia secundária).
Na vasculopatia, a hipóxia, componente fundamental da fisiopatologia da ACF, leva à diminuição da produção de óxido nítrico (NO), o qual é importante regulador do tono vascular, de adesão celular e da formação de trombose.
As consequências são estase, vasoclusão, hipóxia tecidual, trombose, enfarte e fibrose, entre outras alterações crónicas ao nível de vários órgãos. No baço, tal processo de fibrose conduz a redução de dimensões e a uma depressão funcional do órgão, o que corresponde a verdadeira “autosplenectomia”. Os enfartes teciduais traduzem-se na clínica por dor, por vezes intensa, com localização variável.
O processo de falciformação é muitas vezes iniciado e/ou agravado pela diminuição da pressão parcial de oxigénio e pela acidémia.

FIGURA 4 – Vasoclusão na anemia de células falciformes. Adaptado de Embury et al, 1994
Manifestações clínicas
As manifestações clínicas das diferentes formas de doença falciforme são variáveis, constituindo o epifenómeno de anemia hemolítica crónica com episódios de agudização. Em geral, não surgem antes dos 3 meses; as primeiras manifestações podem surgir de forma insidiosa com palidez, icterícia, colúria e esplenomegália, ou aguda (infecção fulminante por exemplo). Nos extremos deste espectro clínico (fenótipo) estão o traço falciforme e a anemia de células falciformes, que descrevemos a seguir.
Traço falciforme
Esta forma clínica, por vezes assintomática, caracteriza-se por manifestações ligeiras e benignas, destacando-se risco aumentado de hematúria, disfunção renal (hipostenúria, compromisso da capacidade de acidificação urinária-pH urinário alcalino) (predomínio de Hb A sobre Hb S). Ambientes de grande altitude (> 3000 metros) ou a prática de exercícios extenuantes (futebol americano, hóquei no gelo, pólo aquático) podem levar a enfartes pulmonares e esplénicos potencialmente fatais.
Anemia de células falciformes ou drepanocitose
As manifestações têm, em geral, início após os 3 meses de idade, coincidindo com a diminuição da Hb F e aumento da HbS, sendo que quanto maior o teor de Hb F menor o risco de falciformação eritrocitária.
Estão classicamente presentes várias alterações além das que surgem nas formas mais benignas (traço falciforme).
Em termos de magnitude e gravidade das manifestações clínicas, a forma heterozigótica Hb Sβ0 é sobreponível à ACF (Hb SS); nos restantes genótipos as manifestações são ligeiras a moderadas.
É importante salientar que todos os órgãos e sistemas podem ser afectados, com maior relevância o respiratório, o cerebrovascular e o renal.
As chamadas crises vasoclusivas agudas (CVO) manifestam-se por sinais e sintomas que dependem da localização específica: sistema respiratório (por ex.: pneumopatia, enfartes pulmonares), osteoarticular (tumefacção simétrica e dolorosa denominada síndroma “mão-pé” ou dactilite drepanocítica, dores nos ossos longos), digestivo (dor abdominal intensa relacionável com enfartes em órgãos abdominais, síndroma da “cintura”), sistema nervoso central (acidente vascular cerebral isquémico/AVC, sobretudo nos territórios das artérias carótida interna, cerebral média e cerebral anterior).
A doença cerebrovascular representa actualmente uma das principais morbilidades da ACF. A incidência de AVC é cerca de 61/10.000 crianças-ano (600 vezes superior à incidência na ausência de ACF) entre os 6 e 18 anos; tal patologia é muito rara durante os primeiros 12 meses de idade. Em regra, cerca de 10% dos doentes irão sofrer um AVC até aos 20 anos de idade. Os chamados enfartes silenciosos (sintomatologia discreta ou ausente, mas com alterações nos exames de imagem) também comprometem o desenvolvimento cognitivo.
A repercussão no sistema respiratório traduz-se mais frequentemente pela síndroma torácica aguda (STA), mais frequente em crianças do que em adultos e a segunda causa mais comum de internamento. O diagnóstico baseia-se no aparecimento de um infiltrado observável na radiografia torácica associado a febre e/ou sintomas respiratórios. Em idades pediátricas, os principais factores desencadeantes são as infecções por S. pneumoniae, Mycoplasma pneumoniae e Chlamydia.
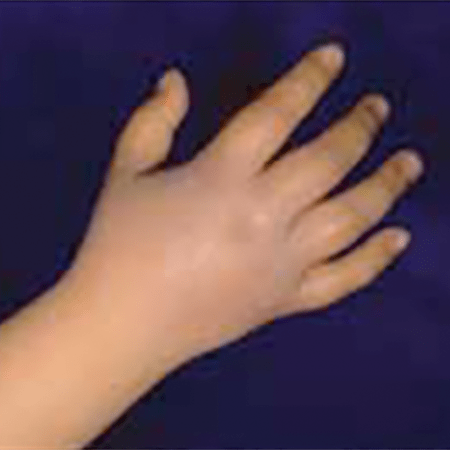
FIGURA 5 – Síndroma mão-pé (dactilite) na ACF. (NIHDE)
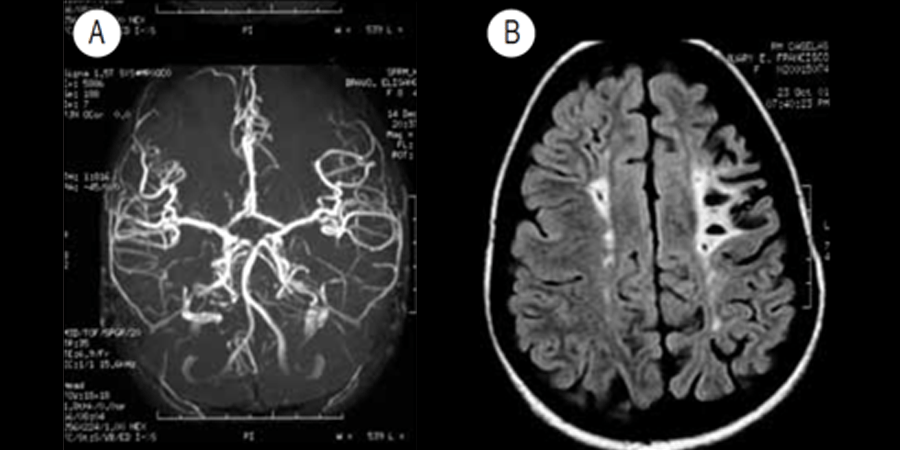
FIGURA 6 – Angiografia (A) e TAC CE (B) – no contexto de ACF: imagens sugestivas de lesões isquémicas por oclusão nos territórios das artérias cerebral anterior e média
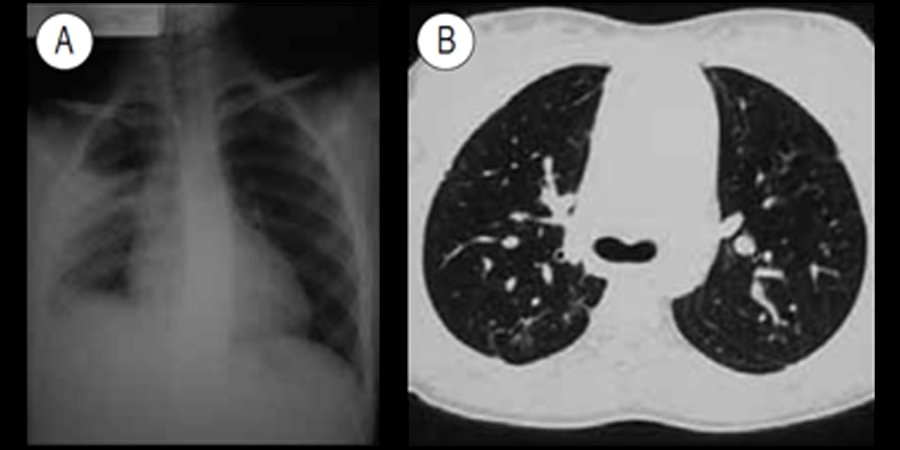
FIGURA 7 – ACF: padrão radiográfico compatível com síndroma torácica aguda. A – Radiografia convencional (infiltrado no hemitórax direito); B – TAC evidenciando alterações fibróticas residuais num adolescente
As denominadas crises de sequestração esplénica, frequentes entre os 6 meses e 3 anos, constituem outro tipo de episódio agudo em que, por causa desconhecida e de forma aguda, se acumulam grandes quantidades de sangue no baço com consequente choque hipovolémico e aumento do volume do baço. Podem também ocorrer no fígado (com a mesma gravidade, mas mais raramente).
As crises hipoplásticas e as crises de híper-hemólise são outro tipo de manifestações típicas. No primeiro caso está, em geral, afectada a série eritróide sendo frequentemente desencadeadas por infecções (designadamente por Parvovirus B19). As crises de híper-hemólise são traduzidas por aparecimento agudo de icterícia (ou por agravamento de icterícia crónica ligeira) e palidez. Estas crises poderão ser agravadas na presença de défice da desidrogenase da glucose-6-fosfato, que deve ser pesquisado de modo sistemático nestes doentes.
Nos adolescentes e adultos jovens poderão surgir úlceras na região maleolar e priapismo; este pode surgir em episódios de duração e periodicidade variáveis.
Existe um risco aumentado de necrose asséptica da cabeça femoral, osteomielite por Salmonella e infecções por Streptococcus pneumoniae e outros agentes capsulados. (Figura 8)
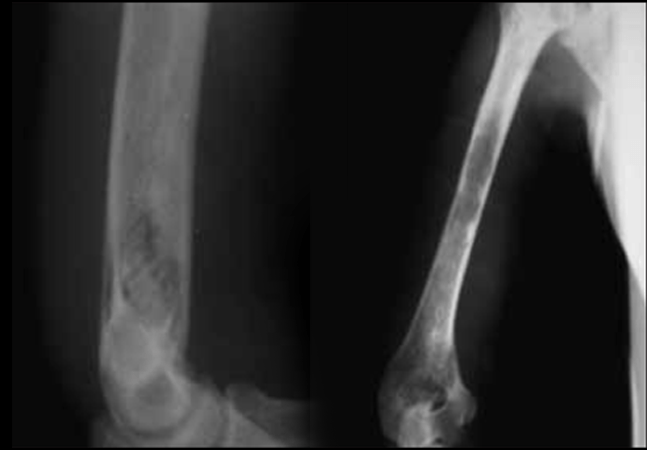
FIGURA 8 – Quadro radiológico de osteomielite do úmero no contexto da ACF. (NIHDE)
A colelitíase, rara na infância, é frequente após os 10 anos. Verifica-se impacte significativo da doença de células falciformes sobre o rim, traduzido por glomerulopatia que, em cerca de 20% dos casos, culmina em insuficiência renal.
Exames complementares
A ACF caracteriza-se por uma anemia normocrómica e normocítica, em geral com valor de Hb entre 7 e 9 g/dL e reticulocitose acentuada (entre 5 e 15%). As formas heterozigóticas em associação com talassémia podem apresentar diminuição do volume globular médio (VGM) e da concentração de hemoglobina globular média (CHGM). No esfregaço do sangue periférico nem sempre são detectadas células falciformes, as quais podem ser evidenciadas pela prova de falciformação; no entanto, tal prova não permite distinguir o estado homozigótico SS do estado de portador heterozigótico ou de outras hemoglobinopatias; o esfregaço permite ainda identificar glóbulos vermelhos nucleados e corpos de Howell-Jolly. A cromatografia permite separar a Hb S das restantes. A proporção de Hb F nas hemoglobinopatias S varia entre 1 e 20%.
O diagnóstico molecular pode ser estabelecido no primeiro trimestre de gravidez por biópsia das vilosidades coriónicas, no segundo trimestre através de amniocentese ou após o nascimento no sangue periférico. Nalguns países é efectuado rastreio neonatal às populações de risco.
O exame da medula óssea mostra sinais de hiperplasia eritróide.
As radiografias do crânio e da coluna vertebral evidenciam córtex estreitado, alargamento do espaço medular; ao nível do crânio é típico o padrão de “crânio em escova”, igualmente observável nas síndromas talassémicas.
O estudo imagiológico por Doppler transcraniano (TCD, sigla em inglês) permite identificar precocemente lesões estenóticas nas artérias carótida interna distal, cerebral média proximal e cerebral anterior e estratificar o risco de AVC isquémico em crianças assintomáticas com drepanocitose entre os 2 e os 16 anos. Nas crianças com achados anormais no TCD, ou naquelas com TCD difícil de avaliar ou condicional, deve ser realizado estudo por ressonância magnética (RM) cerebral para avaliar lesões arteriais ou cerebrais.
Tendo em conta a probabilidade de compromisso renal atrás referido, justifica-se em todos os casos de síndroma falciforme a avaliação anual da microalbuminúria como forma de rastreio, nomeadamente o doseamento de alfa-1 e beta-2 microglobulina.
Tratamento
O tratamento da ACF inclui:
- Medidas gerais: nutrição adequada, prevenção da desidratação, prevenção do arrefecimento corporal, imunizações (designadamente antimeningococócica, anti-Hemophilus influenzae tipo B, anti- pneumoniae conjugada e polissacarídea), evitamento de desportos de moderada a alta intensidade, suporte psicológico e aconselhamento genético;
- Suplementação com ácido fólico;
- Tratamento de crises vasoclusivas (dor intensa em qualquer local do organismo, sendo os ossos os territórios mais frequentemente atingidos) requerendo a administração de analgésicos e promovendo concomitantemente a correcta hidratação endovenosa. Os analgésicos mais frequentemente utilizados são: nas formas mais ligeiras, o paracetamol e/ou anti-inflamatórios não esteróides como o ibuprofeno por via oral ou o cetorolac por via endovenosa; nas formas de dor moderada a grave, a utilização de opiáceos (nomeadamente morfina e fentanil) não deve ser protelada. Salienta-se a importância da analgesia mantida em detrimento da administração SOS; é mais eficaz e reduz o tempo de internamento. A aplicação local de calor constitui factor adjuvante no controlo da crise;
- Prevenção e tratamento de infecções – a profilaxia antibiótica deve ser mantida até aos 5-6 anos, ou indefinidamente em doentes esplenectomizados ou com infecções recorrentes graves. Pode-se utilizar a penicilina (penicilina benzatínica mensal por via intramuscular) nas seguintes doses: crianças com < 10 kg: 300.000 U; 10-25 kg: 600.000 U; > 25 kg: 1.200.000 U. Em alternativa, pode utilizar-se a amoxicilina diária na dose de 20 mg/kg/dia (duas tomas diárias);
- Tratamento da anemia hemolítica crónica – a anemia é bem tolerada na maior parte dos doentes, uma vez que a Hb S tem menor afinidade para o oxigénio, o que facilita a sua libertação ao nível dos tecidos. As transfusões de concentrado eritrocitário têm o objectivo de melhorar a capacidade de transporte de oxigénio e diluir as células falciformes em circulação para melhorar a perfusão microvascular; consegue-se, assim, baixar os níveis da Hb S para valores ≤ 30% da Hb total, ou aumentar a Hb para cerca de 10 g/dL;
- As transfusões têm indicações precisas: Hb < 5 g/dL; nas crises aplástica ou hipoplástica; nos sequestros esplénicos e hepáticos; nos AVC e na sua prevenção, quer primária, quer secundária; nas STA isoladas ou de repetição; nas situações de lesão multiorgânica; e no pré-operatório de intervenções cirúrgicas com anestesia geral. Não deve transfundir-se para valores de Hb superiores a 10 g/dL por risco de hiperviscosidade (nestas situações deve considerar-se a transfusão permuta);
- A esplenectomia está indicada apenas quando as necessidades transfusionais anuais ultrapassam os 200 ou 250 ml/kg, ou no sequestro esplénico grave recorrente; deverá ser protelada, se possível, até cerca dos cinco anos, e seguida de profilaxia antibiótica;
- A hidroxicarbamida (anteriormente designada por hidroxiureia) reduz a frequência de crises vasoclusivas (CVO), síndroma torácica aguda (STA) e de suporte transfusional. Tal fármaco está indicado em doentes Hb SS ou Hb Sβ0 que apresentam:
- 1) ≥ 3 episódios de CVO num período de 12 meses; 2) antecedentes de STA ou anemia sintomática; 3) antecedentes de AVC e contraindicação de suporte transfusional regular. Alguns autores defendem mesmo a utilização em crianças ≥ 9 meses de idade, assintomáticas ou com episódios pouco frequentes de CVO. A dose inicial é de 15-20 mg/kg/dia com incrementos de 2,5-5 mg/kg cada 8 semanas de acordo com toxicidade hematológica até máximo do 35 mg/kg/dia.
- O transplante de células estaminais é o único tratamento curativo, o qual deve ser considerado na presença de, pelo menos, uma das seguintes situações:
- 1) STA com necessidade de internamentos recorrentes; 2) CVO recorrentes; 3) alteração neuropsicológica com RM cerebral anormal; 4) osteonecrose de múltiplas articulações; 5) doença pulmonar estádio I ou II; 6) proteinúria moderada-grave ou taxa de filtração glomerular entre 30-50%; 7) retinopatia proliferativa bilateral com diminuição major da acuidade visual num olho; 8) aloimunização durante suporte transfusional regular.
- Prevenção e tratamento de doença renal crónica – nos casos de microalbuminúria significativa utilizam-se os inibidores da enzima de conversão da angiotensina (IECA);
- A terapêutica génica constitui uma medida promissora de “cura”. O óxido nítrico (NO) tem sido alvo igualmente de investigação para a terapêutica.
As principais indicações para internamento hospitalar são: dor não controlada com analgesia oral, hipertermia (> 40ºC), mau estado geral (choque e desidratação), sinais imagiológicos de infiltrado pulmonar, hiperleucocitose ou leucopénia, respectivamente > 30.000/mmc e < 5.000/mmc, plaquetas < 100.000/mmc, Hb < 5g/dL, e antecedentes de infecção grave.
Prognóstico e prevenção
A sobrevivência dos indivíduos com ACF melhorou drasticamente devido à melhoria das condições socioeconómicas, ao melhor conhecimento da fisiopatologia, à possibilidade de diagnóstico precoce e de prevenção, e ao tratamento das complicações.
Dado que o diagnóstico precoce proporciona a possibilidade de medidas de profilaxia secundária, o aconselhamento genético e a orientação familiar para os portadores do gene da Hb S, o diagnóstico pré-natal e o rastreio no recém-nascido nas áreas do globo com maior prevalência são estratégias de extrema importância para a melhoria do prognóstico.
A prevenção primária do AVC tem sido levada a cabo nalguns centros pela técnica do Doppler transcraniano medindo a velocidade sanguínea na porção terminal da carótida interna, e na porção proximal da artéria cerebral média. Em 30% dos casos evidenciando dados anómalos (lesões estenóticas), existe probabilidade de AVC isquémico dentro do período de 4 anos.
OUTROS DEFEITOS DA HEMOGLOBINA
A Hb E e a Hb C são, respectivamente, a segunda e terceira alteração mais comum em todo o mundo a seguir à Hb S. São ambas raras no mundo ocidental.
A Hb E é comum no Sueste Asiático e no Sul da China e resulta da substituição do aminoácido na posição 26 da cadeia β da hemoglobina (ácido glutâmico por lisina).
A Hb C é encontrada em indivíduos de descendência africana e resulta da substituição do aminoácido na posição 6 da cadeia β da hemoglobina (ácido glutâmico por lisina).
Ambas determinam quadros clínicos benignos e oligossintomáticos (anemia ligeira, células em alvo, reticulocitose discreta). As crianças com Hb SC têm anemia mais ligeira e menor número de crises dolorosas em comparação com as crianças com ACF (Hb SS).
ANEMIA HEMOLÍTICA CONGÉNITA POR HEMOGLOBINAS INSTÁVEIS
Esta entidade (também designada “anemia com corpos de Einz ou Heinz-Ehrlich”) integra quadros diversos de anemia hemolítica intermitente transmitidos de modo autossómico dominante, destacando-se uma característica biológica clássica: o aparecimento de corpos de Heinz nos eritrócitos e reticulócitos após incubação a 37ºC durante 48 horas.
Foram identificadas mais de 200 hemoglobinas instáveis, quase todas decorrentes de mutações de novo, sendo a Hb Koln a mais frequente. A hemólise intensifica-se com episódios febris e com alguns fármacos oxidantes.
O tratamento é de suporte, com suplementos de ácido fólico. Nalguns casos está indicada a esplenectomia. De salientar a necessidade de evitar medicamentos com efeito oxidante e transfusões de sangue.
HEMOGLOBINOPATIAS COM AFINIDADE ANORMAL PARA O OXIGÉNIO
Trata-se de situações em geral transmitidas de modo autossómico dominante.
- Se a afinidade estiver aumentada (contexto de mais de 100 variantes de Hb), a tradução clínica é o défice de oxigenação tecidual, podendo conduzir a eritrocitose compensadora não associada a outra sintomatologia.
Em cerca de 20% dos casos o diagnóstico pode fazer-se mediante a medição da P50 (ver Parte de Perinatologia/Neonatologia), que é baixa (valor normal ~23-29 mm Hg). Os valores de eritropoietina e 2,3- difosfoglicerato são normais. Não é necessário tratamento.
- Se a afinidade estiver diminuída, o resultado poderá ser anemia (com Hb Seattle) ou cianose (com Hb Kansas). As correspondentes variantes da Hb podem resultar de mutações nas cadeias α ou β. A exposição à inalação de oxigénio corrige a cianose, o que não acontece nos casos de metemoglobinémia e de hemoglobinopatia M.
METEMOGLOBINÉMIAS
Patogénese
Em condições normais forma-se continuamente metemoglobina nos eritrócitos a partir da hemoglobina (cerca de 1-2% de Hb está sob esta forma de metemoglobina).
A hemoglobina converte-se em metemoglobina quando o ferro do heme (ferroso na hemoglobina ou Fe2+), uma vez oxidado, passa a férrico ou Fe3+ gerando metemoglobina; esta última é a chamada Hb desnaturada.
A metemoglobina não é um pigmento transportador de oxigénio; assim, a curva de dissociação O2-Hb está também desviada para a esquerda, do que resulta um aumento da afinidade do O2 para a Hb, com défice de libertação de O2 para os tecidos e consequente hipóxia.
Várias enzimas redutoras (metemoglobina-redutases ou diaforases) assegurando a sua retransformação permanente em Hb funcional ou transformação de Fe férrico em Fe ferroso, impedem que aquela percentagem de metemoglobina aumente. A forma principal de diaforases tem por coenzima a NADH (nicotinamida-adenina dinucleótido fosfato) reduzida.
Pode deduzir-se que situações congénitas em que existe défice do referido sistema enzimático, ou anomalias na globina que determinam que os grupos heme existam sempre no estado férrico (formação de Hb anómala designada Hb M), ou adquiridas, em que exista acção directa de compostos oxidantes como nitritos, cloratos, quinonas, originam formação em excesso de metemoglobina.
De salientar que na metemoglobinémia por défice enzimático não existe hemólise.
A clássica cor de chocolate do sangue é notória sempre que a proporção de metemoglobina for superior a 15-20%. Proporções superiores a 70% são potencialmente letais.
Nesta perspectiva, podem ser sistematizadas essencialmente três formas clínicas:
- Congénitas (familiares) decorrentes de deficiência enzimática, de transmissão hereditária recessiva, mais frequente;
- Congénitas associadas a defeito da hemoglobina – Hb M, de transmissão dominante;
- Adquiridas, induzidas pela acção de agentes químicos oxidantes já referidos. Esta última forma clínica é relatada a propósito do diagnóstico diferencial e da actuação terapêutica.
Metemoglobinémia congénita (familiar)
Na maioria dos doentes atingidos por esta doença (transmitida de modo autossómico recessivo e frequente nos índios Navajo) verifica-se défice NADH citocromo b5 redutase ou de diaforase 1. A percentagem de metemoglobina é da ordem dos 40%, não originando, em geral, sintomas; poderá verificar-se cianose ligeira, depressão respiratória ou policitémia compensadora.
Tratando-se de formas assintomáticas, não está indicado qualquer tratamento.
Nas formas sintomáticas a abordagem é semelhante à descrita para as formas tóxicas (adquiridas), adiante referidas a propósito do diagnóstico diferencial.
A electroforese das Hb e o estudo espectrofotométrico contribuem para o esclarecimento diagnóstico.
Metemoglobinémia congénita associada a Hemoglobina M
Existem diversas variantes de Hb M, as quais resultam, como referido antes, de anomalias estruturais da globina (cadeias α ou β).
As formas homozigóticas são letais; nas formas heterozigóticas a percentagem de metemoglobina oscila entre 20% e 30%, a que corresponde clinicamente cianose com PaO2 normal.
Ao contrário do que acontece com a metemoglobinémia por défice enzimático, existe diminuição da afinidade da Hb para o O2 verificando-se, portanto, desvio da curva da Hb-O2 para a direita, permitindo maior distribuição de O2 aos tecidos e explicando, designadamente, que não se verifiquem sintomas respiratórios.
A característica clínica mais chamativa é a cianose verificável a partir dos 4-6 meses de idade; nas variantes de Hb M Saskatoon e Hyde Park (hemoglobinas instáveis) pode verificar-se anemia hemolítica crónica.
A electroforese das Hb e o estudo espectrofotométrico contribuem para o esclarecimento diagnóstico.
Nas formas sintomáticas a abordagem é semelhante à descrita para as formas adquiridas, relatadas a seguir.
Diagnóstico diferencial e tratamento
O diagnóstico diferencial das metemoglobinémias congénitas hereditárias faz-se essencialmente com a metemoglobinémia adquirida (tóxica).
Esta situação resulta da acção de certas drogas e agentes químicos oxidantes que provocam desnaturação da hemoglobina tais como toxinas produzidas por certas enterobacteriáceas em casos de diarreia, nitritos, nitratos (certos aditivos alimentares, fertilizantes), primaquina, derivados da anilina (corantes, certos lápis), sulfonamidas, análogos da vitamina K, benzocaína, etc.. Os recém-nascidos são mais susceptíveis à formação de metemoglobina dado que possuem maior percentagem de hemoglobina F e mais baixo nível de metemoglobina-redutase.
As manifestações clínicas traduzem-se essencialmente por cianose que não responde à administração de oxigénio. Aliás, trata-se duma pseudocianose com coloração da pele descrita classicamente como “mais castanha do que azul”. Tais manifestações somente se verificam se a taxa de Hb reduzida for > 5 g/dL. Se os valores de metemoglobinémia forem > 1,5 g/dL, o sangue evidencia cor castanha (tipo “chocolate”).
O sintomas e sinais (tanto mais exuberantes quanto maior o teor de metemoglobina formada), em presença de pressão arterial de O2 (Pa O2) normal ou elevada, são: ansiedade, cefaleia, tontura e síncope surgem com níveis entre 20-30%, enquanto confusão, prostração, taquicardia e taquipneia surgem com níveis entre 30-50%. Níveis > 70%, que podem ser letais, associam-se a acidose metabólica, arritmia cardíaca, convulsão e coma.
Deve admitir-se a hipótese de metemoglobinémia na presença de uma disparidade entre a saturação arterial em oxigénio medida por oximetria de pulso e por gasometria arterial – “saturation gap/hiato na saturação”. A co-oximetria permite determinar a percentagem das diferentes formas de hemoglobina e estabelecer o diagnóstico de metemoglobinémia.
O tratamento (de urgência) da metemoglobinémia tóxica (adquirida) consiste na administração por via endovenosa de azul de metileno (solução a 1%) na dose de 1-2 mg/kg de peso durante 5 minutos; a dose pode ser repetida em intervalos de 4 horas até máximo de 7 mg/kg. [O azul de metileno está contraindicado nos casos de défice dedesidrogenase da glucose-6-fosfato/ G– 6PD]. Em alternativa: ácido ascórbico na dose de 200-500 mg/dia (efeito mais lento). Nos casos em que não se verifica resposta está indicada exsanguinotransfusão ou oxigenação hiperbárica.
Nota: O azul de metileno e o ácido ascórbico são ineficazes em casos de metemoglobinémia associada a Hb M.
BIBLIOGRAFIA
Adekile AD, Gupta R, Khayat AA, et al. Risk of avascular necrosis of the femoral head in children with sickle cell disease on hydroxyurea: MRI evaluation. Pediatric Blood & Cancer 2019; 66: e27503
Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica 2014; 99: 811-820
Balwani M, Desnick R. The porphyrias: advances in diagnosis and treatment. Blood 2012; 120: 4496-4504
Cao A, Galanello R. Beta-thalassemia. Genet Med 2010; 12: 61–76
Chiruka S, Darbyshire P. Management of thalassaemia. Paediatr Child Health 2011; 21: 353-356
Claster S, Vichinsky EP. Managing sickle cell disease. BMJ 2003; 327: 1151-1555
Coughlin K, Flibotte J, Cahill AM, et al. Methemoglobinemia in an infant after sclerotherapy with high-dose doxycycline. Pediatrics 2019; 143: (2) e20181642; DOI: 10.1542/peds.2018-1642
Cunningham MJ. Update on thalassemia: Clinical care and complications. Pediatr Clin North Am 2008; 55: 447-460
Driscoll MN, Hurlet A, Styles L, et al. Stroke risk in siblings with sickle cell anemia. Blood 2003; 101: 2401-2404
Gillis VL, Senthinathan A, Dzingina M, et al. Guideline development Group. Management of an acute painful sickle cell episode in hospital: summary of NICE guidance. BMJ 2012; 27: 344
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Haridasa N, DeBaun MR, Sanger M, et al. Student perspectives on managing sickle cell disease at school. Ped Blood & Cancer 2019; 66: e27507
Hoffman R, Benz EJ, Silberstein LE, et al (eds). Hematology: Basic Principles and Practice. Philadelphia: Elsevier, 2018
Howard J, Hart N, Roberts-Harewood M, et al. Guideline on the management of acute chest syndrome in sickle cell disease. Br J Haematol 2015; 169: 492–505
Ivankovich DT, Braga JAP, Lanza FC, et al. Lung function in infants with sickle cell anemia. J Pediatr 2019; 207: 252-254
Jayavaradhan R, Malik P. Genetic therapies for sickle cell disease. Pediatr Clin North Am 2018; 65: 465-480
Lichtman MA, Williams WJ (eds). Williams Hematology. New York: McGraw-Hill, 2006
Kayle M, Docherty SL, Sloane R, et al. Transition to adult care in sickle cell disease: A longitudinal study of clinical characteristics and disease severity. Ped Blood & Cancer 2019; 66: e27463
Kassim AA, Galadanci NA, Pruthi S, DeBaun MR. How I treat and manage strokes in sickle cell disease. Blood 2015; 125: 3401-3410
Kliegman RM, Stanton BF, StGeme JW, Schor NF (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2015
Kumar G, Muwaijei AA, Sohal APS, et al. Sickle cell crisis: A crisis of a different sort? Arch Dis Child Educ & Practice 2018; 103: 290-336
Meier ER. Treatment options for sickle cell disease. Pediatr Clin North Am 2018; 65: 427-444
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Nathan DG, Oskin SH, Ginsburg D, Look AT (eds): Hematology of Infancy and Childhood. Philadelphia: Saunders, 2003
Peters M, Heijboer H, Smiers F, Giordano PC. Diagnosis and management of thalassaemia. BMJ 2012; 344: e228
Piel FB, Tatem AJ, Huang Z, et al. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob Health 2014; 2: e80–e89
Rehman HU. Methemoglobinemia. West J Med 2001; 175: 193-196
Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA (eds). Rudolph´s Pediatrics. New York: McGraw-Hill Medical, 2011
Rund D, Rachmilewitz E. Beta -Thalassemia. NEJM 2005; 353: 1135-1146
Smith-Witley K, Thompson AA. Indications and complications of transfusions in sickle cell disease. Pediatr Blood Cancer 2012; 59: 358-364
Taher A, Vichinsky E, Musallam K, et al. Guidelines for the management of non-transfusion dependent thalassaemia (NTDT). Nicosia-Cyprus: Thalassaemia International Federation ed, 2013
Telfer PT. Management of sickle cell disease: outpatient and community aspects. Paediatr Child Health 2011; 21: 357-362
Towerman AS, Hayashi SS, Hayashi RJ, et al. Prevalence and nature of hearing loss in a cohort of children with sickle cell disease. Ped Blood & Cancer 2019; 66: e27457
Willen SM, DeBaun MR. The epidemiology and management of lung diseases in sickle cell disease: lessons learned from acute and chronic lung disease in cystic fibrosis. Pediatr Clin North Am 2018; 65: 481-494
Wong TE, Brandow AM, Lim W, Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood 2014; 124: 3850-3857
Wonke B. Clinical management of beta-thalassemia major. Semin Hematol 2001; 38: 350-359
Zaidi AU, Heeney MM. A scientific renaissance: novel drugs in sickle cell disease. Pediatr Clin North Am 2018; 65: 445-494