Exames complementares
A anamnese e o exame objectivo permitem a suspeita de doença neuromuscular, assim como da localização (segmento da unidade motora provavelmente afectado).
Os exames complementares de diagnóstico mais úteis são a enzimologia muscular (doseamento da fosfocreatinocinase – CPK), o electromiograma (EMG), a biópsia de músculo, a biópsia de nervo, as provas terapêuticas (como a prova do edrofónio), e os estudos de genética molecular. Refira-se ainda o estudo metabólico, os exames imagiológicos (TAC e RM), e a avaliação cardíaca (ecocardiografia).
O doseamento da CPK poderá ser útil na diferenciação entre doenças musculares primárias e neuropatias. No recém-nascido, um aumento de CPK poderá indicar distrofia muscular congénita ou distrofia miotónica congénita; no lactente e criança mais velha colocam-se as hipóteses de distrofia muscular congénita, miosite, distrofia muscular progressiva (mesmo em fase pré-sintomática), miopatias congénitas e distrofia miotónica infantil. Nos rapazes no 3º ano de vida com atraso na aquisição ou alterações da marcha, o doseamento de CPK é importante, e caso seja elevado (> 10.000 UI/L) há indicação para realizar estudo genético, dispensando-se o EMG ou a biópsia muscular.
O EMG permite diferençar qual o segmento da unidade motora afectado, sendo especialmente útil para o rápido diagnóstico de atrofia muscular espinhal tipo I (AME I – doença de Werdnig-Hoffman), de neuropatia (distinguindo a neuropatia desmielinizante da axonal, sendo fundamental o registo da velocidade de condução nervosa), e de doença da placa motora (a estimulação repetitiva do músculo induz fatigabilidade progressiva).
A biópsia de músculo (com microscopia óptica, electrónica ou com estudo imuno-histoquímico) permite o diagnóstico dos vários tipos de doenças musculares. Na distrofia muscular congénita, distrofia muscular progressiva e distrofia miotónica congénita, a microscopia óptica comprova a distrofia, sendo necessário o estudo imuno-histoquímico para uma classificação mais completa (estudo da presença de merosina ou de distrofina e sarcoglicanos, com recurso a técnicas de Western Blotting para análise quantitativa). Igualmente importante para caracterizar várias miopatias congénitas e para o diagnóstico de miopatia inflamatória.
A biópsia do nervo confirma a hipótese de neuropatia, classificando-a (por exemplo: desmielinizante ou hipomielinizante).
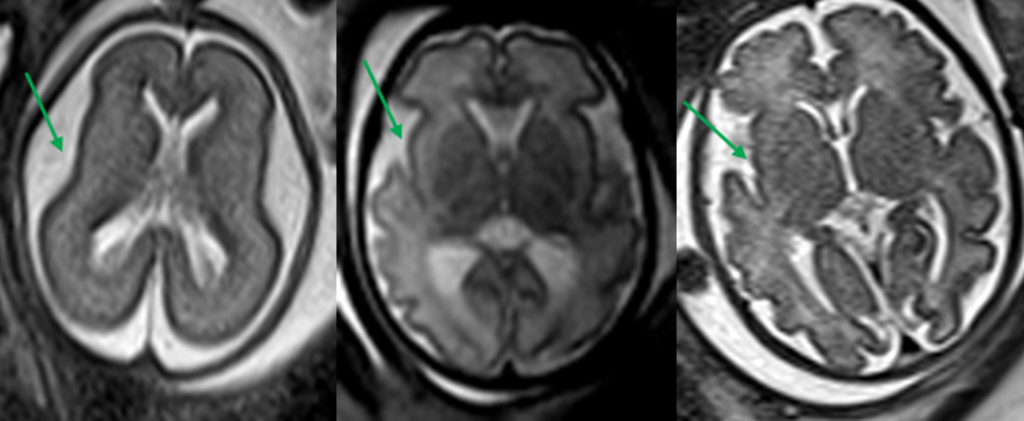
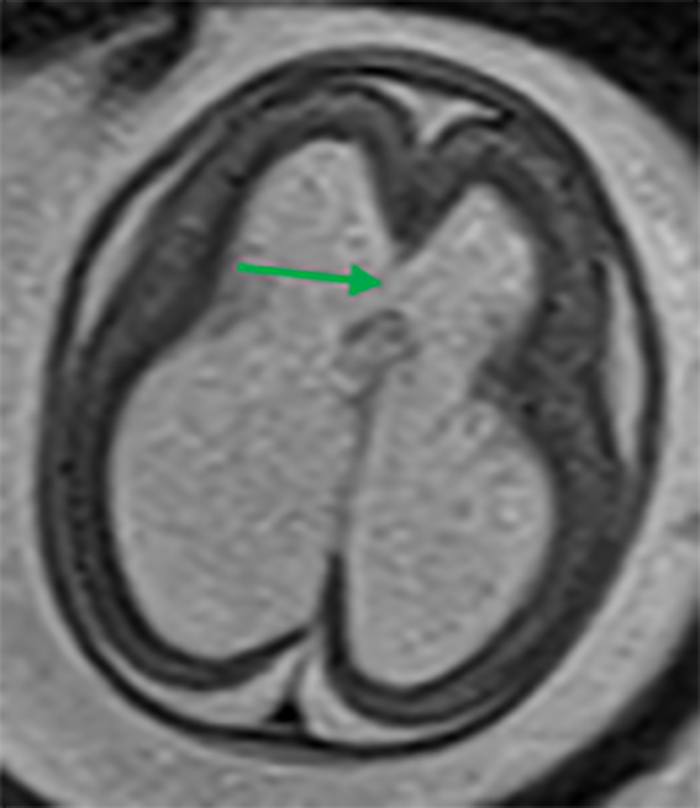
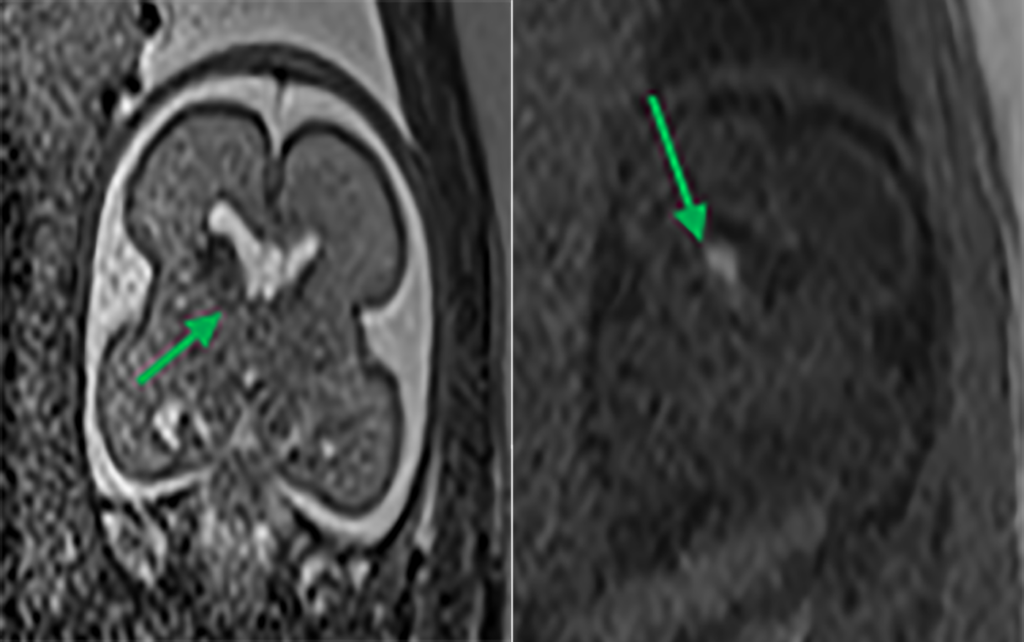
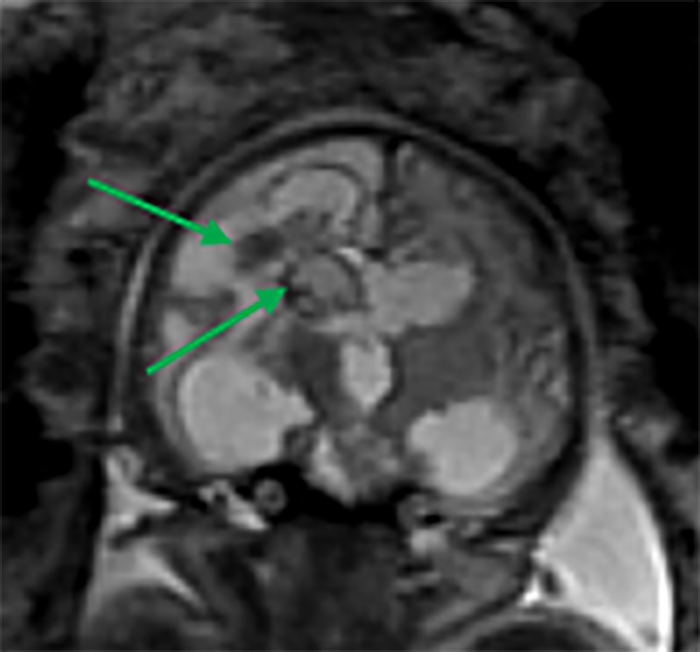
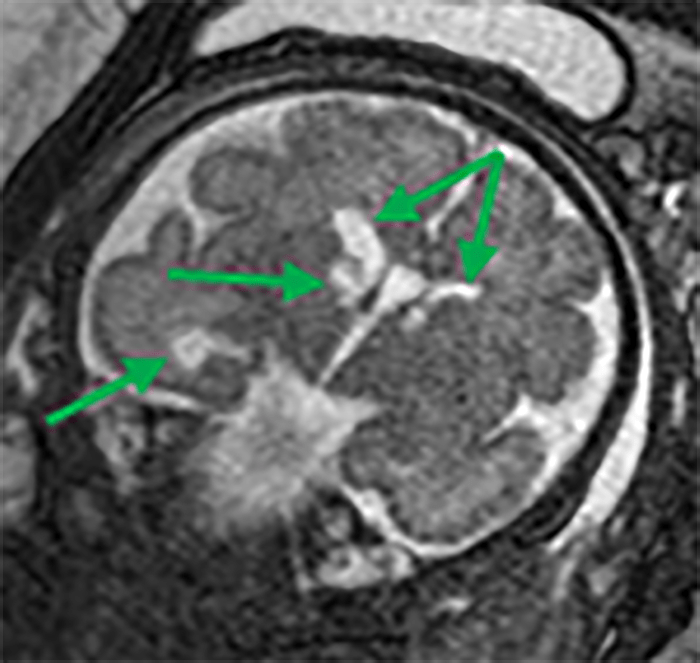
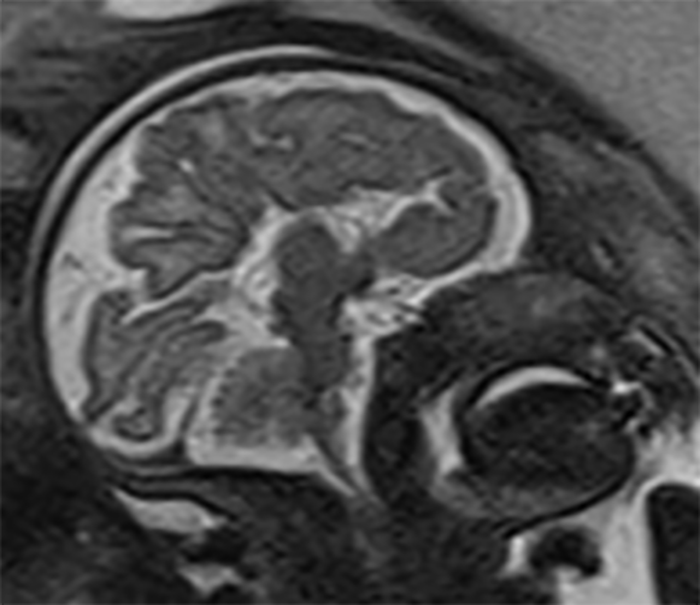
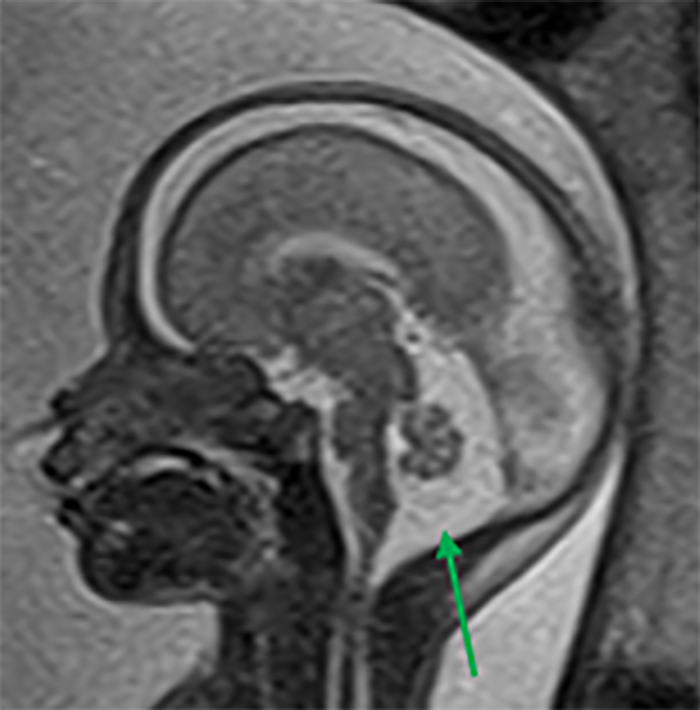
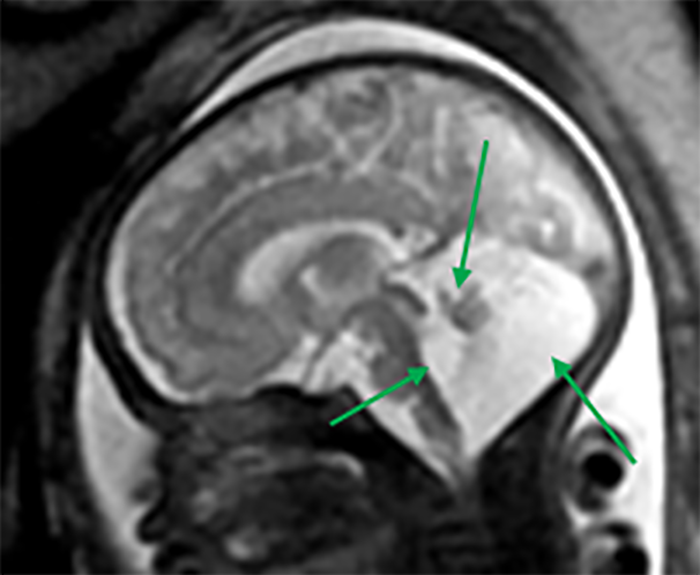
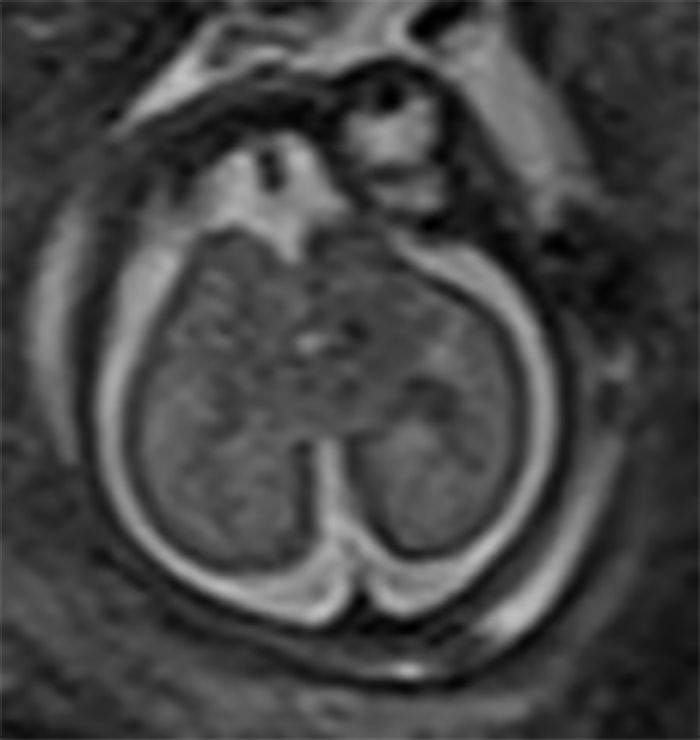
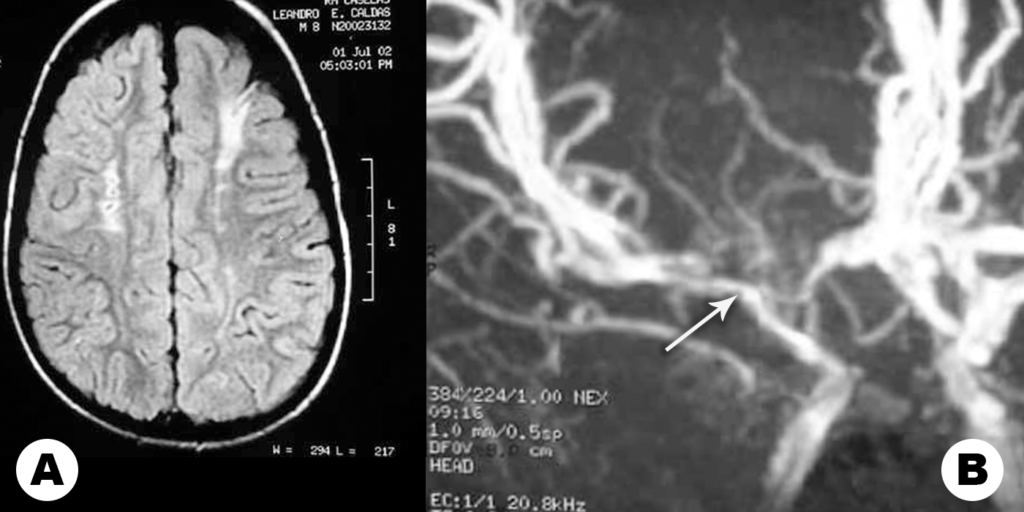
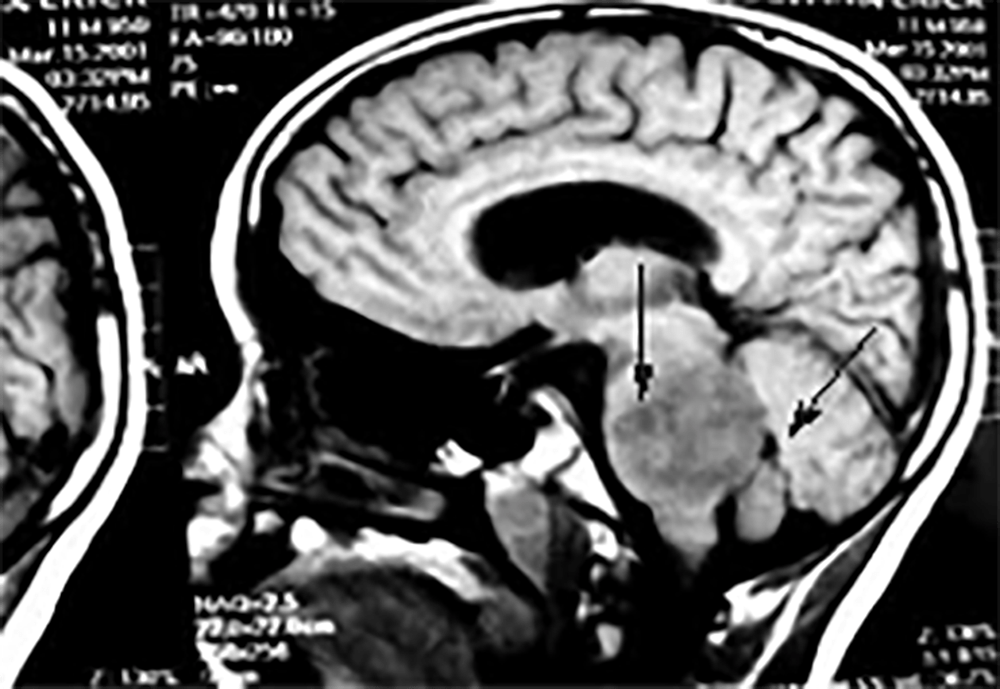
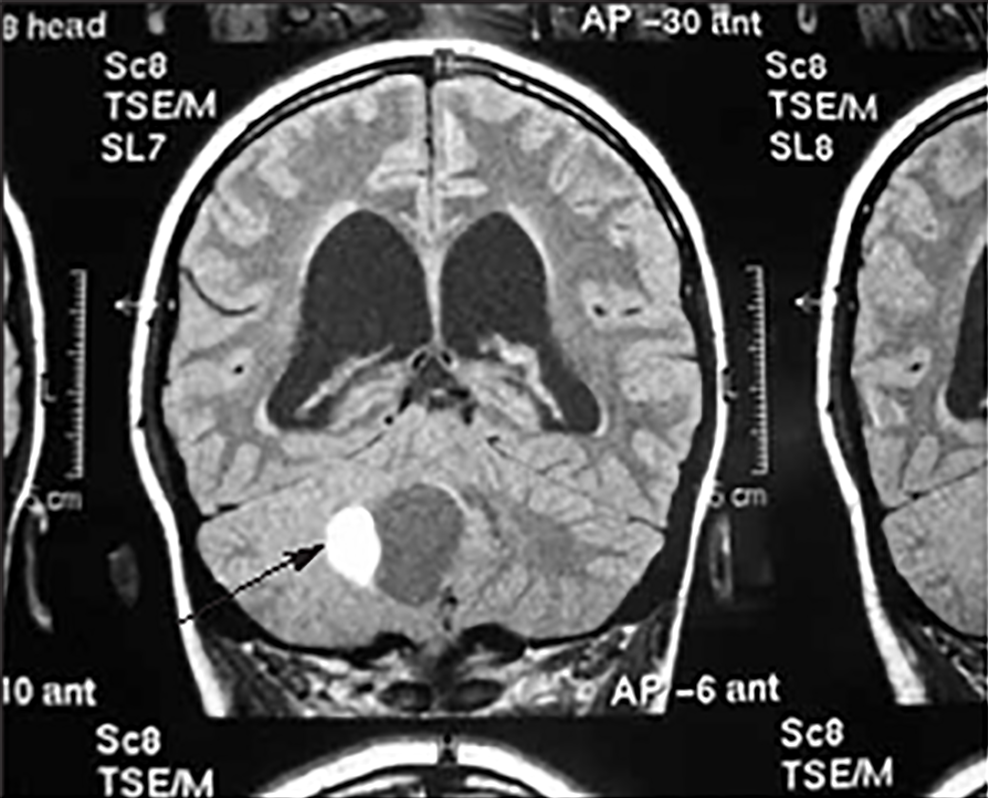
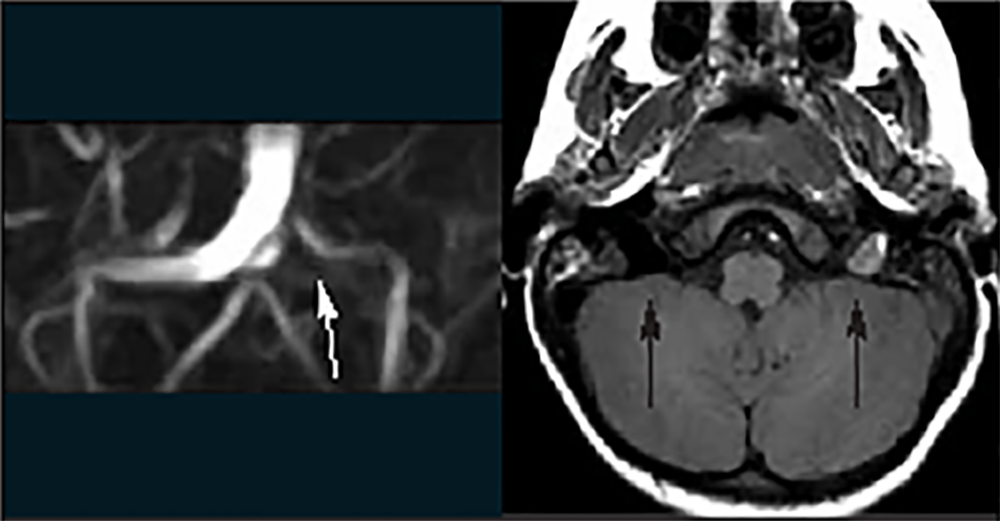
A RM – CE poderá ser útil nas citopatias mitocondriais (alteração de sinal dos núcleos da base e da substância branca), na distrofia muscular congénita com afecção do SNC (defeitos de desenvolvimento cortical e atrofia cerebelosa) e na distrofia miotónica congénita (áreas de possível gliose cerebral).
O estudo metabólico (lactato, piruvato, amónia, aminoácidos, ácidos orgânicos, entre outros) deve ser realizado se existir suspeita de doença metabólica.
A avaliação cardiológica deve ser feita na suspeita de doença neuromuscular que se associe a cardiopatia (miocardiopatia ou disritmias), como a distrofia muscular progressiva – de Duchenne ou de Becker, algumas mitocondriopatias, glicogenoses, algumas miopatias congénitas e distrofia miotónica.
Os estudos de genética molecular, realizados em centros especializados, são essenciais para a confirmação diagnóstica de algumas doenças neuromusculares e sua classificação mais exacta (classificação que, com os progressos rápidos da ciência, se desactualiza a breve trecho).
Os estudos de genética molecular são essenciais para a confirmação diagnóstica das doenças neuromusculares genéticas. Estes estudos são particularmente úteis para distinção das diferentes formas de distrofias musculares, de miopatias congénitas, de miopatias mitocondriais e de polineuropatias sensitivo-motoras (agrupadas com a designação genérica de CMT- proveniente do epónimo doença de Charcot-Marie-Tooth, que designa grande parte das neuropatias genéticas). Igualmente nos casos suspeitos de distrofinopatia pela verificação de elevação do CPK em jovens pré-escolares do sexo masculino, com hipertrofia dos gémeos/ retração do tendão de Aquiles, ligeiro atraso no início da marcha, com ou sem antecedentes familiares de doença muscular, está indicado o estudo inicial do gene DMD.
Saliente-se que os painéis NGS atualmente disponíveis não identificarão as doenças neuromusculares genéticas devidas à variação do número de cópias, sendo necessário para estas métodos de diagnóstico próprios (como ocorre na maioria dos casos de Distrofia Muscular de Duchenne/ Becker, distrofia fascio-escápulo-umeral, ou na distrofia miotónica, por exemplo).
Tratamento
A abordagem terapêutica destas doenças consiste sobretudo em métodos paliativos, como a reabilitação motora e a cirurgia ortopédica, tentando minorar os défices motores apresentados pelos doentes. A abordagem terapêutica do recém-nascido com grave compromisso motor e respiratório é delicada, constituindo um problema ético. Nas patologias em que pode haver compromisso da função respiratória, esta deve ser avaliada periodicamente, iniciando, logo que se justifique, programa de ventilação (inicialmente não invasiva, e intermitente, como o BIPAP nocturno); se existirem dificuldades alimentares há que ponderar a gastrostomia. A restante patologia associada (cardiológica, oftalmológica, pedopsiquiátrica, otorrinolaringológica) deverá ser avaliada pelo especialista respectivo por indicação do médico responsável.
Alguns tipos de doença neuromuscular progressiva têm terapêutica farmacológica específica (por exemplo: várias modalidades de imunomodulação e medicamentos colinérgicos na miastenia grave, e corticóides na distrofia muscular de Duchenne).
Uma nova etapa no tratamento das doenças neuromusculares genéticas começa agora. Em 2017 foi aprovada a primeira terapia genética para Atrofia Muscular Espinhal (nusinersen). As terapias aprovadas ou em avaliação através de ensaios clínicos podem ser assim sumarizadas, referindo-se apenas as principais:
- Terapêuticas de interferência no mRNA:
- exon skipping com oligonucleotidos antisense para promover a restauração da reading frame em diversos estudos nomeadamente em doentes com Distrofia Muscular de Duchenne com deleções que causam disrupção da reading frame
- modificadores do splicing do pre mRNA com oligonucleotidos antisense terapêuticas já aprovadas para Atrofia Muscular Espinhal (nusinersen e risdiplam)
- supressão do nonsense promove a leitura do mRNA através de um codão stop prematuro, aumentando a síntese da proteína (ataluren, já aprovado para Distrofia Muscular de Duchenne com mutações nonsense)
- Terapia génica: é realizada a transdução do gene através da sua inclusão num vector viral (AAV- vírus adeno-associados) administrado endovenosamente (onasemnogene abeparvovec, já aprovado para Atrofia Muscular Espinhal, outros em avaliação em ensaios clínicos)
A integração do indivíduo com doença neuromuscular no seu meio é um desafio multidisciplinar, envolvendo diferentes parceiros (assistente social, professores, entre outros), e a obtenção de ajudas técnicas (cadeiras de rodas, coletes ortostáticos, computadores, etc.).
Formas clínicas
De acordo com o segmento da unidade motora afectado, são descritas sucintamente as doenças neuromusculares mais frequentes e/ou mais típicas em clínica pediátrica.
1. Doenças do corno anterior medular
Atrofia muscular espinhal (AME)
Importância do problema: trata-se de uma doença degenerativa (por mecanismo apoptótico) dos cornos anteriores medulares e dos núcleos motores de alguns pares cranianos.
Constitui a segunda doença neuromuscular mais frequente (a seguir à distrofia muscular de Duchenne), com uma incidência de 1/20.000 recém-nascidos.
Etiologia: a AME é uma doença genética autossómica recessiva associada em 95% dos casos à deleção homozigótica do exão 7 do gene SMN-1 (Survival Motor Neuron, de localização telomérica), no braço longo do cromossoma 5 (5q11q13). A gravidade do fenótipo relaciona-se com o número de cópias existentes do gene SMN-2 (idêntico ao SMN-1, mas situado no centrómero); é menos grave se houver muitas cópias presentes, justificando-se assim a variabilidade fenotípica.
Patologia: existe atrofia muscular neurogénica (desnervação) ou secundária.
Clínica e evolução: variam de acordo com a idade de início e a gravidade do envolvimento motor:
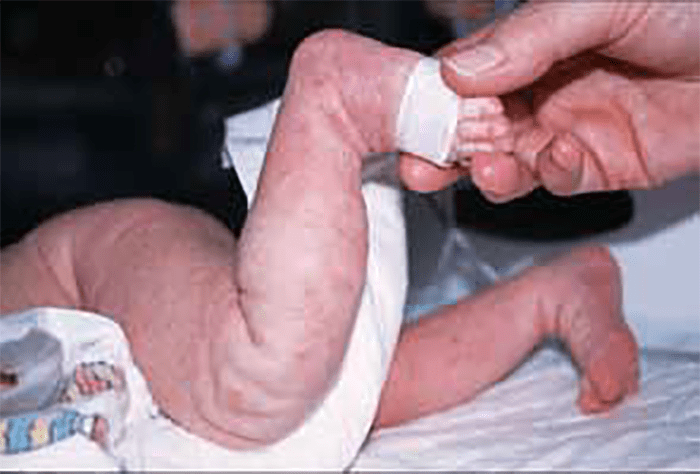
- AME – 1 (Doença de Werdnig-Hoffmann). É a causa mais frequente de hipotonia neuromuscular no recém-nascido e no lactente. Os sinais clínicos têm início antes dos 6 meses de idade, não adquirindo a criança a capacidade de se sentar sem apoio. Os movimentos fetais são escassos. As manifestações iniciais são: hipotonia progressiva, parésia de predomínio proximal, arreflexia e fasciculações da língua. A afecção dos músculos intercostais e bulbares leva a compromisso respiratório, com insuficiência e infecções respiratórias graves, sendo estas a causa de mortalidade, ocorrendo geralmente antes dos 2 anos de idade.
- AME – 2 (Forma Intermédia). Inicia-se entre os 6 e 12 meses de idade, com hipotonia e parésia (sobretudo dos membros inferiores). A criança consegue sentar-se sem apoio, embora não adquira a marcha. Há um progressivo envolvimento dos membros superiores e dos músculos respiratórios, com compromisso respiratório na segunda década de vida (causa de morte).
- AME – 3 (Doença de Kugelberg-Welander). Tem início após os 18 meses de idade, com aquisição da marcha (embora com dificuldades associadas). Há diminuição da força das cinturas pélvica e escapular. Poderá haver perda da marcha na segunda década de vida.
Diagnóstico: é confirmado pelo EMG, biópsia de músculo e estudo de genética molecular.
Poliomielite
Etiopatogénese e clínica: a infecção pelo poliovírus tipos 1-3 (enterovírus) é hoje pouco comum nos países desenvolvidos e designadamente em Portugal, o que se explica pelo sucesso dos programas de imunização.
O período de incubação oscila geralmente entre 8-12 dias (com variações entre 5 e 35 dias).
Descrevem-se as seguintes formas clínicas:
- Forma assintomática (mais de 90% dos casos);
- Doença minor ou não paralítica (cerca de 5% dos casos). As manifestações clínicas são: febre, mal-estar, odinofagia e vómitos surgindo cerca de 4 dias após exposição ao vírus; a evolução é favorável com cura espontânea;
- Forma paralítica (cerca de 0,1% dos casos) ocorrendo com uma sequência de manifestações idênticas às da doença minor;
- Por sua vez, a poliomielite paralítica integra 3 síndromas distintas relacionadas com os territórios do SNC mais intensamente afectados:
- bulbar acompanhada de paralisias dos músculos faciais, da mastigação, respiratórios, etc. em função dos centros afectados;
- polioencefalite em que se verifica compromisso dos centros superiores do encéfalo; podem surgir convulsões e coma, para além de paralisia espástica e sinais de irritação meníngea;
- espinhal: é esta forma que é paradigmática da afecção do corno anterior e que justifica a inclusão da poliomielite neste capítulo de doenças neuromusculares.
Para além de fasciculações e espasmos, salienta-se a particularidade da paralisia flácida assimétrica, sobretudo das áreas proximais do membro inferior de um lado (um só músculo ou grupos de músculos), podendo posteriormente outro membro (superior) também ser atingido. A fase de paralisia tem duração variável com recuparação ou sequelas, o que depende do grau de lesão neuronal. A paralisia dos membros inferiores pode associar-se a disfunção vesical ou dismotilidade intestinal. Se forem afectados os segmentos espinhais cervicais e torácicos, pode surgir insuficiência respiratória.
Tratamento: o tratamento é sintomático, sendo que não existe tratamento específico antivírico. Na fase aguda estão contraindicados procedimentos cirúrgicos e injecções intramusculares.
De referir que as estirpes de vacina viva podem originar infecções fatais em crianças com agamaglobulinémia ou imunodeficiência combinada.
2. Polirradiculoneuropatias
Síndroma de Guillain-Barré (SGB)
Definição e importância do problema: trata-se de uma polirradiculoneuropatia desmielinizante inflamatória aguda, levando a paralisia progressiva após infecção ou imunização.
A SGB tem uma prevalência de 1-4/100.000, afectando, em geral, as crianças com idade superior a 2 anos. Ocorre insuficiência respiratória em 25% dos casos, sendo necessária ventilação artificial. A mortalidade é cerca de 2-3% na criança, sendo superior no adulto (até 15%).
Etiopatogénese: observa-se lesão do neurónio motor (raiz e nervo periférico) com desmielinização, presença de linfócitos e de macrófagos, mediada por mecanismo auto-imune (presença de auto-anticorpos anti-gangliósido – GM1 e GM1b). Uma infecção ou imunização que leve a uma alteração das populações de células T supressoras, e de linfócitos T e B que reconhecem antigénios do sistema nervoso, poderá estar na génese do SGB. A infecção desencadeante tem geralmente etiologia vírica (VEB, CMV, VHA, VHB, vírus da varicela-zóster, vírus do sarampo e da rubéola, Influenza A e B, Coxsackie e Echovirus), embora possa ser bacteriana (Campylobacter jejuni e Mycoplasma). As vacinas anti-rábica ou anti-influenza também se associam a SGB.
Clínica e evolução: a redução gradual da força e as parestesias são as queixas iniciais. A avaliação neurológica revela uma paralisia generalizada, essencialmente simétrica, geralmente distal (embora possa ser proximal ou mista), com carácter ascendente (sequencialmente: membros inferiores, membros superiores, tronco, e face) e arreflexia generalizada. A paralisia dos músculos respiratórios com necessidade de ventilação mecânica é uma complicação da SGB.
Caso haja ataxia e oftalmoplegia é provável tratar-se da síndroma de Miller-Fisher (SGB com afecção dos pares cranianos). Há sinais de disfunção dos nervos autonómicos tais como hipotensão, taquicardia, hipertensão e arritmia cardíaca; pode verificar-se igualmente disfunção do esfíncter vesical. Após o início dos primeiros sintomas pode haver agravamento no período de 10 a 30 dias.
Diagnóstico: o exame do LCR revela dissociação albumino-citológica (hiperproteinorráquia com contagem celular < 10 células/mm3). A electrofisiologia revela diminuição das velocidades de condução nervosa sensitiva e motora, compatível com desmielinização. O diagnóstico diferencial deve ser feito com as neuropatias periféricas (tóxicas e infecciosas), a poliomielite (sobretudo a vacinal), mielopatia aguda por compressão medular (tumor, trauma, abcesso), esclerose múltipla, doença muscular (polimiosite, miopatia mitocondrial) e doença da placa neuromuscular (miastenia gravis).
Prognóstico: a recuperação é, em geral, completa, havendo sequelas neurológicas em 5-25% dos doentes. Pode haver recorrência de SGB em 3% dos casos. Os factores de mau prognóstico são: maior gravidade do défice motor; maior período desde o início da doença até ao início da recuperação, e EMG com sinais de desnervação.
Tratamento: a instabilidade clínica obriga a internamento hospitalar com monitorização contínua dos parâmetros vitais; poderá ser necessário entubação para ventilação imediata. A abordagem terapêutica actual baseia-se na administração de imunoglobulina (2 g/kg) por via intravenosa (dose total), em 2 dias – com resultados sobreponíveis à plasmaferese.
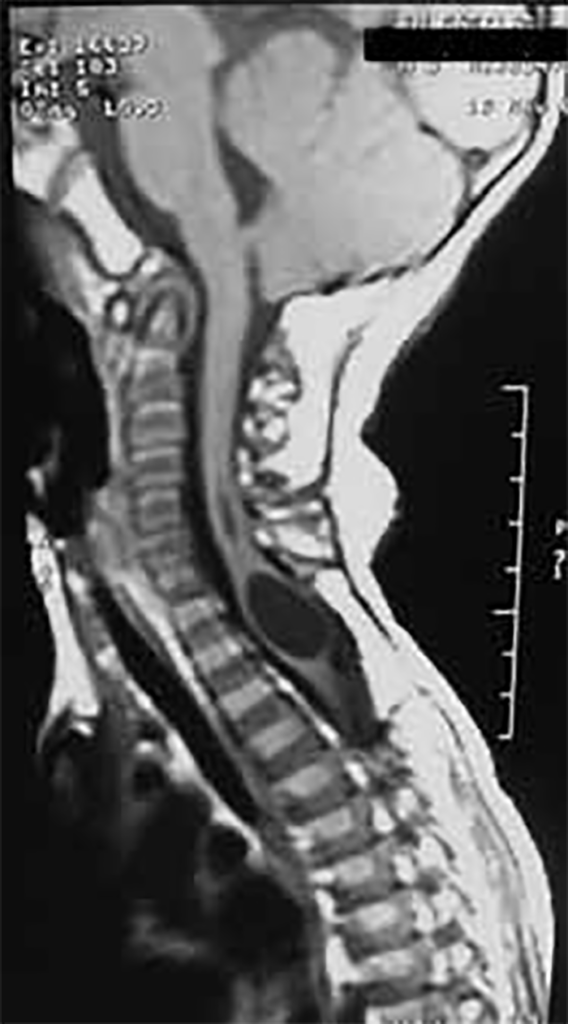
Neuropatias hereditárias sensitivo-motoras (CMT/Charcot-Marie-Tooth)
Importância do problema: as neuropatias hereditárias sensitivo-motoras são o grupo de doenças degenerativas do sistema nervoso periférico mais comuns na criança (40% das neuropatias crónicas).
Etiopatogénese e clínica: a degenerescência da bainha de mielina e/ou axónios leva a uma amiotrofia paralítica distal com arreflexia, envolvendo inicialmente os membros inferiores. Os avanços na genética molecular contribuiram para uma melhor compreensão destas doenças.
A classificação actual combina critérios electromiográficos (demielinizante versus axonal) com padrões de transmissão genética: dominante e desmielinizante (CMT1), dominante e axonal (CMT2), recessiva (CMT4).
- CMT1A (dominante, desmielinizante, duplicação do gene PMP22 no cromossoma 17); pode surgir na criança depois dos 3 anos de idade.
- CMT4 (recessiva, múltiplos genes descritos) (o epónimo Déjerine- Sottas era anteriormente usado para algumas destas neuropatias); o quadro clínico tem início na infância precoce com paralisia predominantemente distal, arreflexia, por vezes hipertrofia de troncos nervosos, associando-se a não aquisição ou perda da marcha.
- Uma forma congénita mais grave (neuropatia congénita hipomielinizante, também com vários genes identificados) pode ainda determinar insuficiência respiratória e compromisso dos músculos bulbares.
Diagnóstico: o EMG é fundamental, revelando redução na velocidade de condução nervosa. A biópsia de nervo realiza-se actualmente com menor frequência, tendo vindo a ser substituída pelos estudos de genética molecular.
Paralisia de BELL
Definição e etiopatogénese: a paralisia de Bell é uma paralisia aguda do nervo facial, unilateral, não associada a outras neuropatias cranianas ou a disfunção do tronco cerebral.
Surge em todas as idades abruptamente, cerca de 2 semanas após uma infecção vírica (mais frequentemente por vírus Herpes simplex tipo 1), mas também em associação a Mycoplasma ou Borrelia. De tal resulta neuropatia desmielinizante do VIIº nervo craniano.
Trata-se dum processo de mecanismo imune, secundário à agressão infecciosa inicial. No período neonatal, a paralisia facial pode resultar de compressão traumática do nervo facial por forceps.
Clínica, tratamento e prognóstico: verifica-se no lado afectado parésia da hemiface, sulco nasogeniano menos marcado, comissura labial mais aproximada da linha média e impossibilidade de aproximação das pálperas (lagoftalmo por paralisia orbicular).
Existe diminuição da sensibilidade gustativa dos 2/3 anteriores da língua em cerca de 50% dos casos. No RN a assimetria da mímica facial pode raramente ser causada por ausência congénita do músculo depressor angular oris.
Por vezes verifica-se hipertensão arterial. Como há impossibilidade de aproximação das pálpebras do lado afectado (trata-se duma paralisia facial periférica) pode surgir conjuntivite ou ceratite secundária, implicando cuidados especiais (protecção do globo ocular) com penso oclusivo, a definir pelo oftalmologista.
A prednisolona oral (1 mg/kg/dia) durante 7 dias, iniciada nos primeiros 3-5 dias da evolução poderá contribuir para processo de melhoria mais rápida. A fisioterapia está indicada nos casos arrastados.
O prognóstico é favorável com recuperação espontânea em cerca de 90% dos casos, a qual, no entanto, pode verificar-se em 2-3 meses.
3. Doenças da junção neuromuscular
Estas doenças, raras em Pediatria, integram três tipos:
Síndroma miasténica congénita (não autoimune)
Pode manifestar-se desde o nascimento (raramente), ou durante a infância. Diferentes defeitos ao nível pré-sináptico, sináptico ou pós-sináptico levam a défices distintos (nos receptores colinérgicos, na acetilcolinesterase, etc.). Existem diferentes padrões de transmissão genética (autossómica recessiva ou autossómica dominante) e várias mutações descritas.
O diagnóstico de síndroma miasténica congénita deve ser considerado quando há hipotonia com choro fraco (num recém-nascido ou lactente), fatigabilidade afectando a musculatura ocular, bulbar e dos membros, existência de familiar com quadro clínico semelhante, resposta electromiográfica alterada com a estimulação repetitiva e doseamento de anticorpos anti-receptores de acetilcolina (ACh) negativo. A resposta à terapêutica com medicamentos colinérgicos é geralmente insuficiente.
Miastenia gravis com início juvenil (autoimune)
Caracteriza-se por apresentação aguda de fraqueza muscular nos membros, com fadiga crescente ao longo do dia e envolvimento bulbar (dificuldade na mastigação, na deglutição e na fonação) e ocular (ptose palpebral bilateral e oftalmoplegia). Associa-se a outras doenças autoimunes, sobretudo a hipotiroidismo. A investigação deverá incluir uma prova terapêutica (com edrofónio, ou com neostigmina); o doseamento de anticorpos anti-receptores de ACh; o EMG (com estimulação repetitiva de um nervo motor, obtendo-se potenciais cada vez menos amplos, com aumento do tempo de latência pela fatigabilidade muscular); e a TAC torácica (para pesquisa de timoma). A abordagem terapêutica inclui fármacos colinérgicos, corticoterapia, imunossupressores, gamaglobulina endovenosa, plasmaferese e, por vezes, a timectomia.
Miastenia neonatal transitória
Trata-se duma forma transitória no recém-nascido, filho de mãe com miastenia gravis (ocorrendo em 15% dos casos); manifesta-se nas primeiras 48 horas de vida com sinais miasténicos acentuados (SDR, hipotonia, actividade motora diminuta, reacção fraca ou ausente, dificuldade na deglutição) que duram enquanto houver anticorpos anormais no sangue e músculo. Não existe risco da miasteria grave mais tarde.
O tratamento é sintomático (assistência respiratória, alimentação com sonda gástrica, etc.), incluindo em geral a administração de colinérgicos.
4. Doenças musculares
As doenças musculares constituem um conjunto heterogéneo de patologia afectando primariamente o músculo, na sua maioria transmitidas geneticamente.
Distrofias musculares progressivas
O termo distrofia significa crescimento anormal e deriva do Grego trophe, que corresponde a alimento ou nutrição.
Uma distrofia muscular distingue-se de todas as outras doenças neuromusculares por 4 critérios obrigatórios: miopatia; base genética; evolução progressiva; e degenerescência e morte das fibras musculares em diversas fases da doença.
As distrofias musculares progressivas heredofamiliares são caracterizadas anátomo-patologicamente por alteração do músculo (fibras musculares necrosadas, com sinais de regeneração, hialinizadas, com mistura de fibras atróficas e hipertróficas, e ainda proliferação de colagénio e adipócitos na zona da lesão das fibras musculares), o que se traduz na clínica pela ocorrência de pseudo-hipertrofia dos gémeos e miocardiopatia).
Para além da distrofia muscular de Duchenne e de Becker, a que se dá ênfase como formas de distrofia muscular progressiva, cabe referir ainda a distrofia facio-escápulo-umeral (apenas citada).
- Distrofias musculares progressivas de Duchenne (DMD) e de Becker (DMB). A DMD é a doença neuromuscular hereditária mais comum, com padrão de transmissão recessiva ligada ao cromossoma X. Tem uma incidência aproximada de 17 por 100.000 recém-nascidos. A DMB tem uma menor incidência (cerca de um terço), mas igual prevalência devido à maior longevidade nesta última.
Pelo facto de a proteína implicada na etiopatogénese ser a distrofina, estas doenças também se denominam distrofinopatias. O gene da distrofina, localizado no braço curto do cromossoma X (Xp21), apresenta habitualmente deleção, sendo possível demonstrá-lo em 60-70% dos casos na DMD, e em 90% dos casos na DMB.
A distrofina localiza-se nas membranas celulares dos miócitos, encontrando-se também no SNC. Cerca de 30% dos casos deve-se a novas mutações. As mulheres portadoras são geralmente assintomáticas, embora raramente possa haver manifestações ligeiras a moderadas (por lionização desigual, mosaicismo X0/ XX, ou cromossoma X anómalo).
Na DMD a distrofina está ausente, e na DMB há produção de distrofina, embora em menor quantidade ou com menor peso molecular.
As manifestações clínicas iniciais da DMD têm início entre os 2-4 anos (por vezes mais cedo), com pseudo-hipertrofia dos gémeos, sinal de Gowers e marcha miopática. Aos 6-7 anos surge envolvimento da cintura escapular, e entre os 9-12 anos há perda da marcha autónoma. No final da segunda década de vida ou início da terceira há insuficiência respiratória e cardíaca, conduzindo à morte. Na DMB os sintomas iniciam- se entre os 6-7 anos (ou mais tarde); a perda da marcha nem sempre acontece.
Ocorre défice cognitivo, em geral ligeiro, em 30% dos casos de DMD, e em 10% dos casos de DMB. A cardiomiopatia observa-se em mais de metade dos doentes com distrofinopatia, afectando sobretudo a parede póstero- lateral do ventrículo esquerdo e levando a valvulopatias e arritmias.
O diagnóstico baseia-se fundamentalmente na genética molecular e na biópsia muscular (nos 20% dos casos em que não se encontra a mutação). Em determinados casos, o doseamento de CPK (geralmente > 10.000 UI/L, sendo normal < 160) poderá dar o seu contributo. É possível diagnóstico pré-natal.
O tratamento com corticóides – prednisolona ou deflazacort – com início aos 5-6 anos (ainda com a massa muscular conservada), em esquema intermitente, parece reduzir a velocidade da progressão da doença, podendo atrasar em 1 a 3 anos a utilização da cadeira de rodas, e a progressão da cifoscoliose. O transplante de mioblastos e a terapia génica encontram-se em investigação. Relativamente a este último tópico, cabe referir estudos sofisticados (englobando vectores adenovíricos e fragmentos da molécula da distrofina/microdistrofina, de resultados ainda não conclusivos) com o objectivo de restaurar a expressão da distrofina.
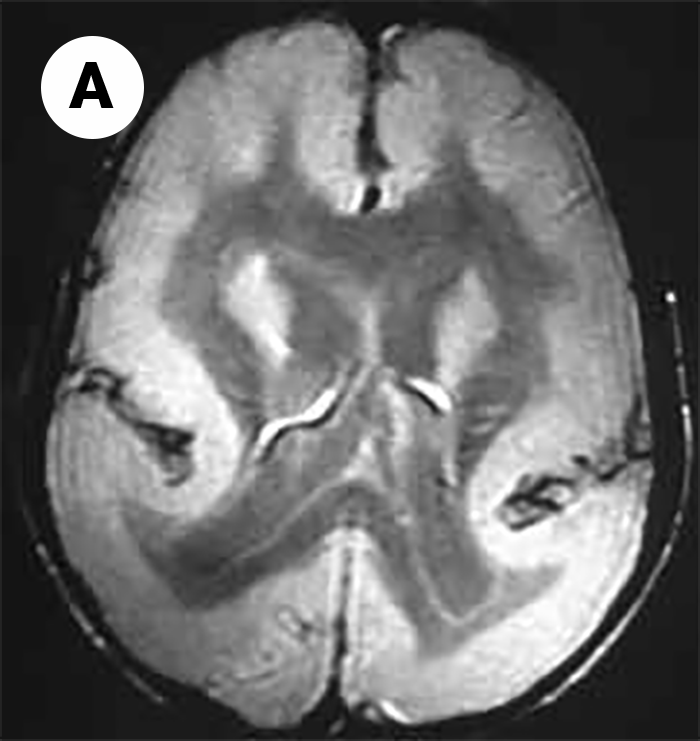
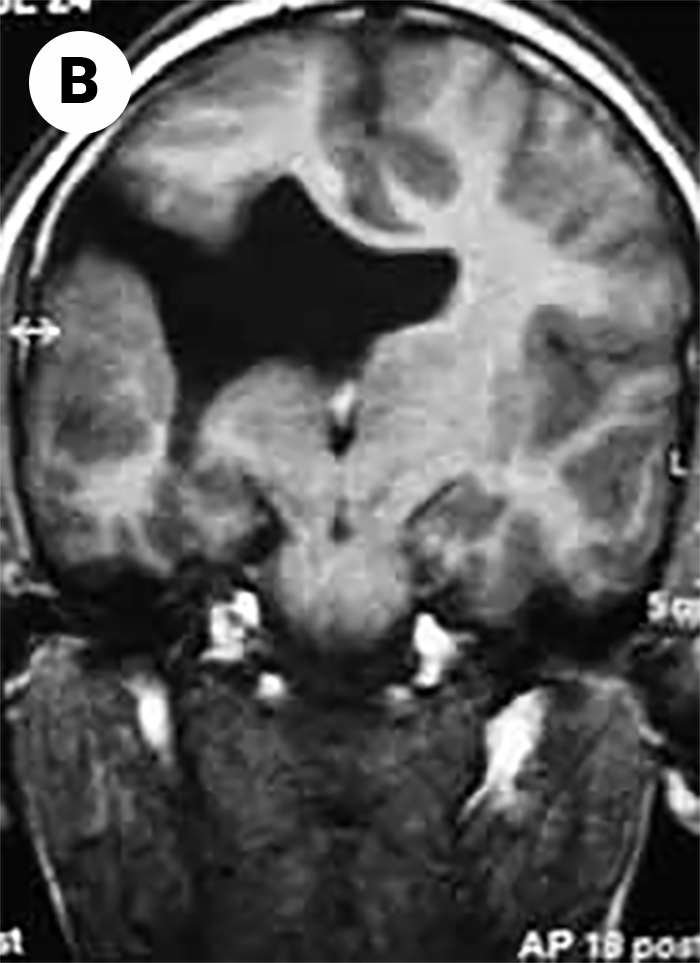
Distrofias musculares congénitas (DMC)
A designação de DMC pode considerar-se confusa, pois todas as DM são geneticamente determinadas (transmissão AR é a regra).
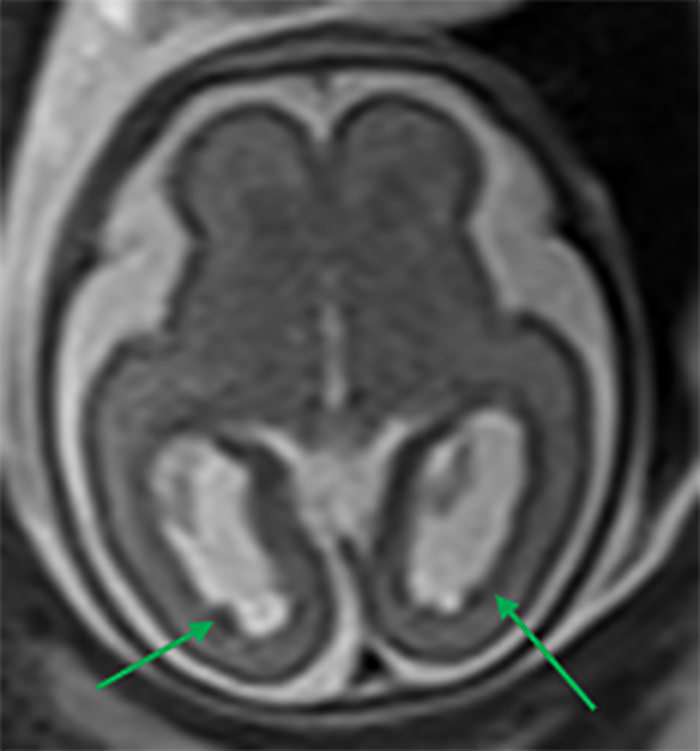
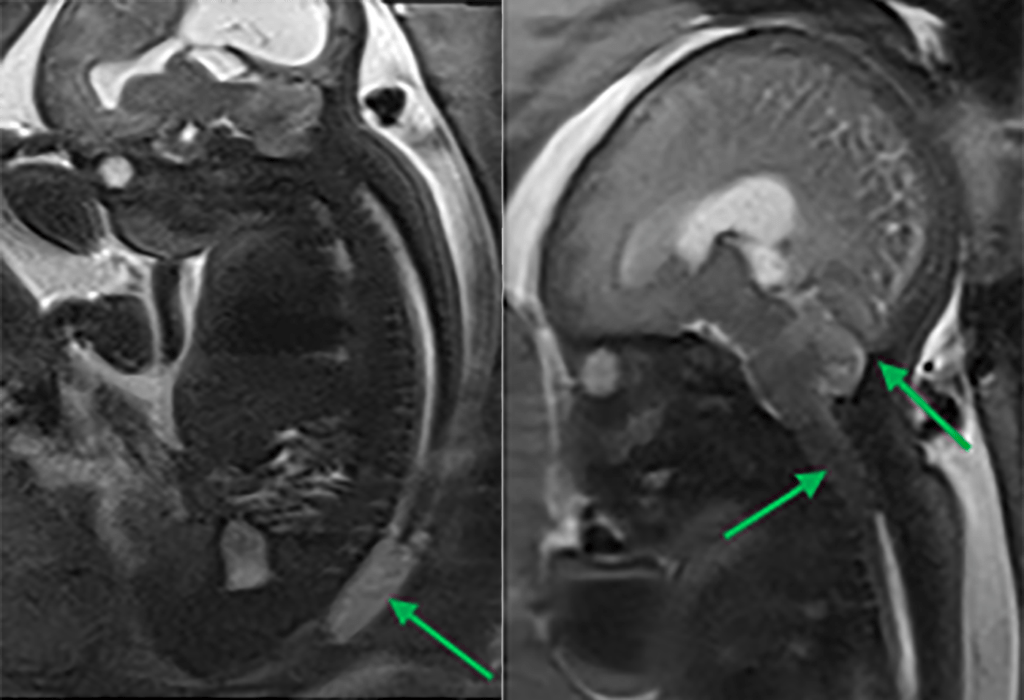
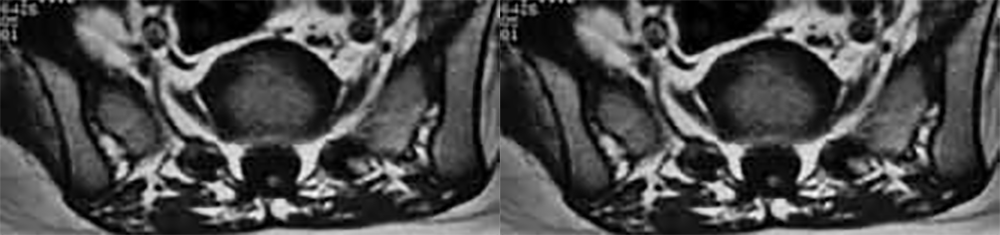
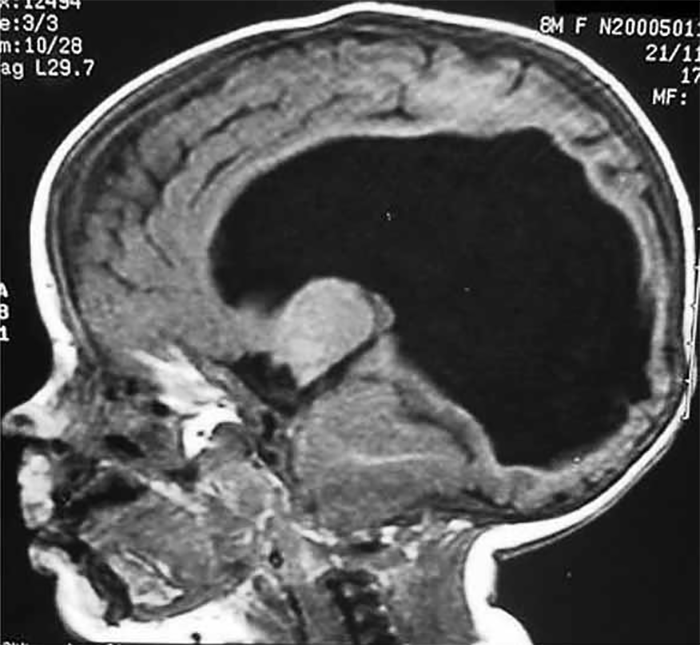
Estas situações correspondem a um grupo heterogéneo de doenças degenerativas primárias e progressivas do músculo esquelético, com início no período intrauterino, ou até ao primeiro ano de vida. A incidência é cerca de 1/60.000 nascimentos, e a prevalência é de 1/100.000 habitantes. Várias DMC têm sido identificadas com base nas características clínicas, patológicas e genéticas: deficiência de merosina ou laminina alfa 2, tipo Ullrich (genes do colagénio 6A), com «rigid spine» (gene SEPN1), síndromas oculocerebromusculares (múltiplos genes) e outras.
No tipo mais frequente (no Japão, Alemanha, Escandinávia e Turquia) a seguir à DMD foi identificado defeito genético no locus 8q 31-33; e em certas formas clínicas, mutações em genes essenciais para a migração do neuroblasto no SNC, como o POMT1. É o designado por “tipo de Fukuyama”.
As DMC caracterizam-se por hipotonia, paralisia com predomínio proximal, arreflexia, retracções tendinosas, frequente afecção dos músculos respiratórios e dificuldade alimentar. Nalguns casos há afecção do SNC, com anomalias estruturais encefálicas, cardiomiopatia e microcefalia.
O diagnóstico baseia-se na clínica, no doseamento de CPK (com ligeiro/moderado aumento), na biópsia muscular e na genética molecular (diagnóstico definitivo).
Histologicamente ocorrem alterações distróficas musculares (fibras com calibre variável, com necrose e proliferação de tecido intersticial), podendo a imuno-histoquímica revelar a presença, ou não, de merosina (alfa-2 laminina).
A evolução clínica é em geral lentamente progressiva ou estática, podendo haver compromisso respiratório (com envolvimento do diafragma), levando à morte.
Outras distrofias musculares
Estão de longa data descritos na literatura neurológica doentes ou famílias com um fenótipo semelhante a DMD ou intermédio entre DMD e DMB, e um padrão de transmissão de doença recessiva ou dominante. Este grande grupo de distrofias musculares foi progressivamente individualizado com base em estudos de genética molecular. Utiliza-se habitualmente a sigla LGMD (limb-girdle muscular distrophy).
O tipo LGMD1 corresponde às formas dominantes que têm em geral uma apresentação mais tardia e um curso menos grave. O tipo LGMD2 designa as formas recessivas. Os subtipos classificam-se com letras (exemplos LGMD2A-calpainopatia, LGMD2B-disferlinopatia, LGMD2C-alfa-sarcoglicanopatia, etc.).
As distrofias musculares de tipo recessivo têm frequentemente um início na primeira infância. São relativamente frequentes, evidenciando pseudo-hipertrofia dos gémeos e cardiomiopatia. Algumas crianças têm uma apresentação «pseudo-metabólica» com episódios de mioglobinúria associados com esforço ou doenças infecciosas.
Miopatias congénitas
As miopatias congénitas são doenças primárias do músculo, na sua maioria de transmissão genética (com vários padrões), e manifestação precoce. Na base desta patologia estão diversos genes implicados em anomalias do processo de diferenciação da célula mesodérmica indiferenciada.
A evolução é lentamente progressiva ou estática. A histopatologia revela anomalia estrutural muscular, com variações no tamanho e número de fibras e/ou presença de inclusões evidenciadas por microscopia electrónica. Na sua origem haverá provavelmente uma anomalia do desenvolvimento e maturação das fibras musculares.
Geneticamente estão descritos diferentes loci implicados.
Como manifestações clínicas referem-se hipotonia, hiporreflexia, amimia facial, micrognatia e palato ogival. A CPK pode estar normal ou moderadamente aumentada; e o EMG revela potenciais motores polifásicos de baixa amplitude. A biópsia muscular associada à microscopia electrónica e a genética molecular confirmam o diagnóstico.
As miopatias congénitas mais bem caracterizadas são:
- Miopatia central core
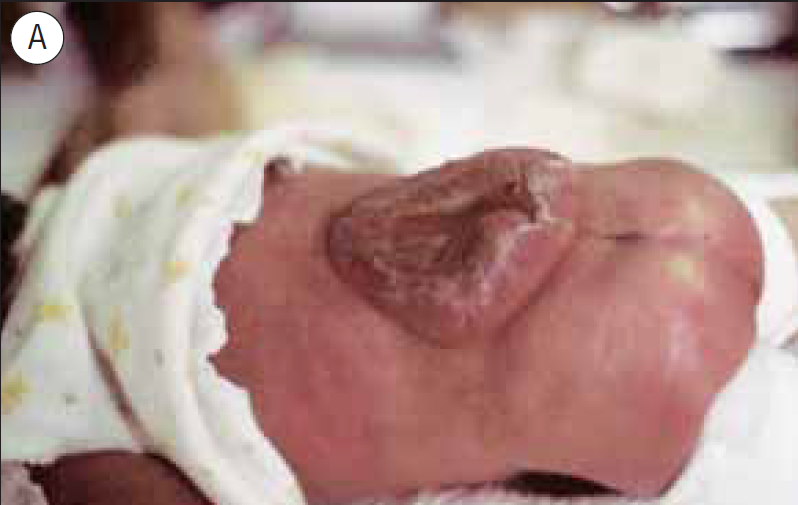
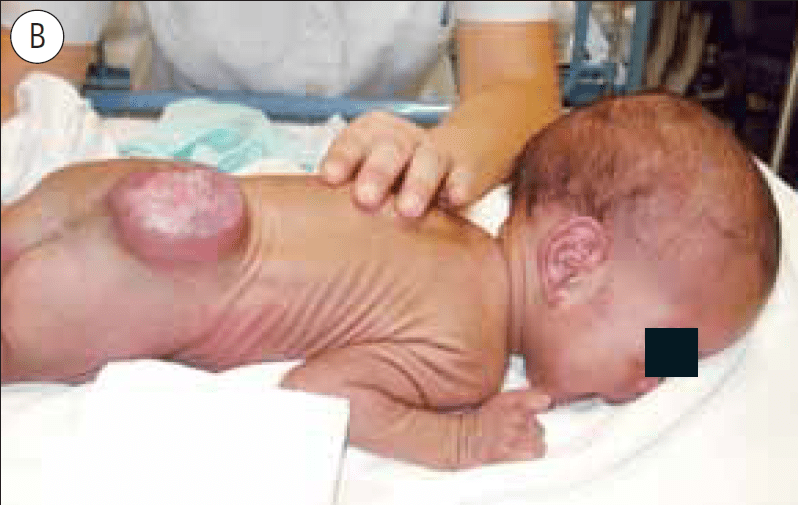
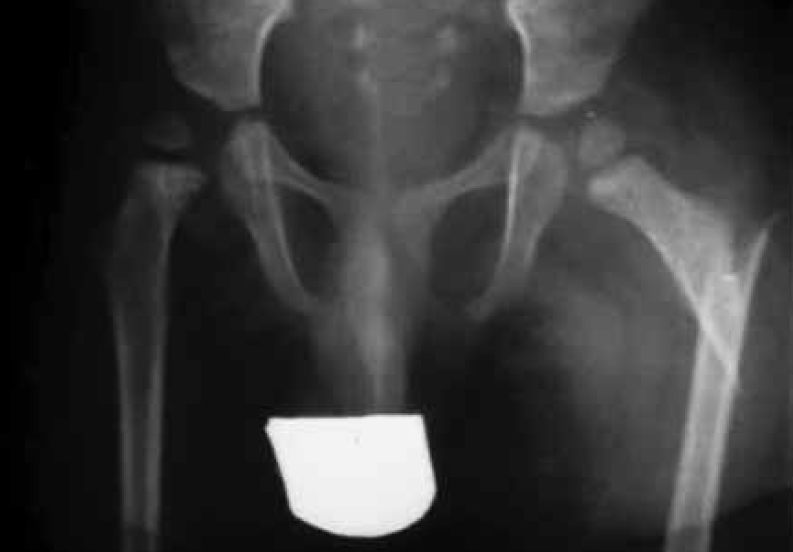
Pelo exame anátomo-patológico identificam-se, nas fibras musculares tipo I, áreas centrais desprovidas de enzimas oxidativas. Há hipotonia neonatal e deformidades-luxação congénita da anca, cifoscoliose e contracturas dos dedos da mão em flexão.
Estão descritos padrões de transmissão dominante, recessiva e formas esporádicas. Este tipo de miopatia evidencia susceptibilidade à hipertermia maligna.
- Miopatia nemalínica
Histologicamente observam-se estruturas em forma de filamento (rods), compostas por a-actinina e desmina (dos discos z). Há heterogeneidade fenotípica, apresentando a forma mais grave hipotonia neonatal e paralisia proximal, amimia, dificuldade alimentar e respiratória, dismorfismos craniofaciais e envolvimento cardíaco. Os quadros menos graves têm um início mais tardio. O padrão de transmissão pode ser autossómico recessivo ou autossómico dominante. São conhecidas várias mutações genéticas afectando uma proteína muscular específica.
- Miopatia centronuclear
A microscopia revela miotúbulos fetais dispostos centralmente na fibra muscular, sugerindo um atraso na maturação do sistema sarcotubular. Há várias apresentações possíveis: na forma ligada ao cromossoma X (locus Xq28) a sintomalogia clínica é muito grave, com insuficiência respiratória e dificuldade alimentar após o parto; as formas autossómicas (recessivas ou dominantes) são menos graves, com variabilidade fenotípica. A sinomatologia é muito grave, com insuficiência respiratória, dificuldade alimentar, ptose palpebral, oftalmoplegia e ocasional envolvimento do SNC (com convulsões e défice cognitivo).
Doença miotónica (Doença de Steinert)
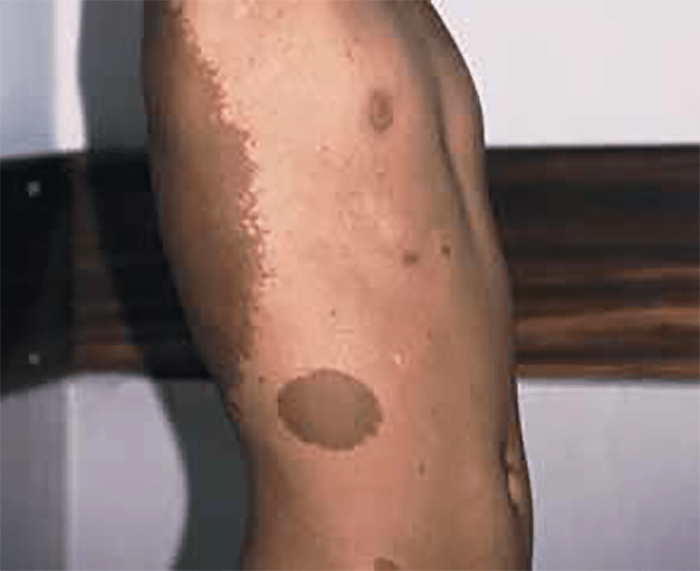
Com uma incidência de 1/30.000 na população geral e, de transmissão AD, é a segunda distrofia muscular mais comum nos EUA, Europa e Austrália. Não somente a musculatura estriada está afectada, mas igualmente e musculatura lisa do aparelho digestivo, útero e coração. Pode haver endocrinopatia, imunodeficiência e cataratas.
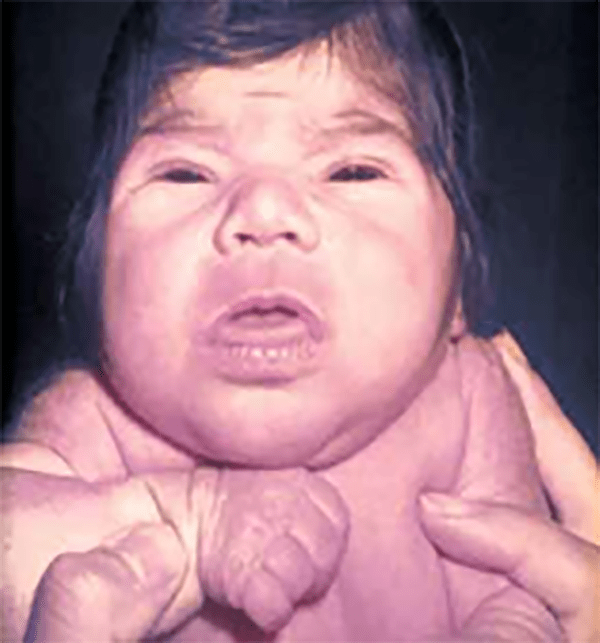
As manifestações iniciais aparecem no período pré-natal (artrogripose múltipla ou polidrâmnio) ou neonatal com hipotonia, fácies miopática, palato ogival, dificuldade alimentar e respiratória (com necessidade de ventilação); a mortalidade é elevada (25% dos doentes) por insuficiência respiratória. As crianças que sobreviveram ao 1º ano podem apresentar défice cognitivo e “fraqueza” facial, (lábio superior em V invertido) com melhoria evidente da função muscular até à 2ª ou 3ª década de vida, altura em que se instala um quadro de miopatia progressiva com défice cognitivo. A miotonia só se observa após o período neonatal. A mãe da criança é doente, podendo não manifestar os sintomas exuberantes.
A biópsia muscular sugere deficiente maturação muscular (fibras pequenas, pouco diferenciadas com padrão muito semelhante ao da miopatia miotubular). A genética molecular contribui para o diagnóstico, revelando a expansão da repetição do tripleto CTG na análise da mutação do gene DMPK (gene da miotonina-locus 19q13.3). Nesta doença há o fenómeno de antecipação que consiste numa maior precocidade no início da doença, e num aumento da gravidade da mesma nas gerações seguintes.
Miosites
A inflamação do tecido muscular ou miosite (pós-infecciosa) é uma situação aguda e transitória, possivelmente mediada imunologicamente, e desencadeada por uma infecção vírica (Enterovirus, Echovirus, Coxsackie B, Influenza A e B, VEB, HSV, Varicella-zoster, entre outros). O quadro clínico consiste em mialgias intensas (geralmente nos gémeos), com dor à palpação dos músculos envolvidos, e impotência funcional. A terapêutica é sintomática, sendo esta situação auto-limitada. As miosites de origem bacteriana ou parasitária são muito raras nos países desenvolvidos.
NOTA: Sugere-se a consulta do Glossário Geral relativamente aos termos Artrogripose e Miotonia.