Definição e nomenclatura
Discrania (ou alocefalia) define-se como anomalia da dimensão e/ou da forma do crânio, congénita ou adquirida, de patogénese diversa, com um espectro variado de manifestações.
A normocrania (ou normocefalia) engloba as situações de normalidade da forma e dimensões do crânio, assim como as respectivas variantes dismórficas consideradas fisiológicas.
Na literatura científica relacionada com a Dismorfologia, existem ainda as designações de deformidades cranianas, dismorfias e de dismorfismos, compreendendo as discranias e as variantes.
Adoptando a classificação de A. Galdó e M. Cruz, são considerados três grandes grupos de discranias: as macrocefalias, as microcefalias e as craniossinostoses.
Como exemplos de normocranias citam-se: mesocefalia, dolicocefalia, braquicefalia e hiperbraquicefalia.
Semiologia
Com interesse semiológico para o diagnóstico diferencial das dismorfias cranianas, e para a caracterização de situações-limite, importa recordar a noção de índice cefálico.
O índice cefálico (IC) obtém-se dividindo o resultado do diâmetro cefálico transversal (DCT) pelo do diâmetro cefálico ântero-posterior (DCAP), multiplicando este quociente por 100: IC = DCT/DCAP x 100.
Os valores traduzindo normalidade de dimensões oscilam entre 70 e 90.
Assim, IC < 75 corresponde a dolicocefalia; IC entre 75 e 79 corresponde a mesocefalia; IC entre 80 e 85 corresponde a braquicefalia; e IC > 85 corresponde a hiperbraquicefalia.
Para as medições pode utilizar-se um instrumento simples (~ pinça curva de abertura larga para adaptação ao crânio, envolvendo-o, articulada com ponteiro deslizando ao longo de régua de leitura acoplada, indicando entre 80 e 170 mm: craniómetro. (Figura 1)
Com interesse semiológico para o diagnóstico diferencial das dismorfias cranianas, e para a caracterização de situações-limite, importa recordar a noção de índice cefálico.
O índice cefálico (IC) obtém-se dividindo o resultado do diâmetro cefálico transversal (DCT) pelo do diâmetro cefálico ântero-posterior (DCAP), multiplicando este quociente por 100: IC = DCT/DCAP x 100.
Os valores traduzindo normalidade de dimensões oscilam entre 70 e 90.
Assim, IC < 75 corresponde a dolicocefalia; IC entre 75 e 79 corresponde a mesocefalia; IC entre 80 e 85 corresponde a braquicefalia; e IC > 85 corresponde a hiperbraquicefalia.
Para as medições pode utilizar-se um instrumento simples (~ pinça curva de abertura larga para adaptação ao crânio, envolvendo-o, articulada com ponteiro deslizando ao longo de régua de leitura acoplada, indicando entre 80 e 170 mm: craniómetro. (Figura 1)
Como notas semiológicas importa ainda salientar:
- a importância da avaliação seriada do perímetro cefálico – com ou sem dismorfia – relacionando-o com outros parâmetros como o peso, idade e estatura;
- perante história de prematuridade e/ou muito baixo peso de nascimento (inferior a 1.500 gramas) deve relacionar-se o perímetro cefálico com a idade pós-concepcional e não com a idade pós-natal (consultando tabelas próprias).

FIGURA 1. Craniómetro
Desenvolvimento do crânio
Na criança, o crânio é uma estrutura que mantendo a rigidez necessária à sua protecção, permite o enorme crescimento do cérebro em tal grupo etário.
A calote craniana, de origem membranosa, é constituída a partir da 6ª semana de gestação pela união de centros de ossificação; por sua vez, a membrana mesenquimal passa a integrar dois folhetos: o externo que origina o pericrânio, e o interno que origina a dura-máter, com poder osteogénico.
Cerca da 23ª semana de gestação está formada a calote craniana, com ossos separados por áreas não ossificadas: 1) fontanelas (anterior ou bregmática, posterior ou lambdóide, e ântero-laterais ou esfenoidais); e 2) suturas (sagital ou interparietal, coronal/transversal ou parietofrontal, metópica ou interfrontal, lambdóide ou parieto-occipital e esfenoparietal); a metópica funde-se até aos 2 anos.
Na data de nascimento os ossos estão justapostos e unidos por tecido fibroso. Relativamente às fontanelas, no recém-nascido após gestação de termo e em condições de normalidade, somente é notória a fontanela anterior; de salientar que o encerramento desta fontanela não é acompanhado de obliteração das suturas.
A fontanela posterior, de escassas dimensões, poderá ser palpada no RN pré-termo ou em casos associados a atraso de ossificação de diversas etiologias, nomeadamente hipotiroidismo. As restantes somente são demonstráveis através de radiografia. (Figura 1-A)
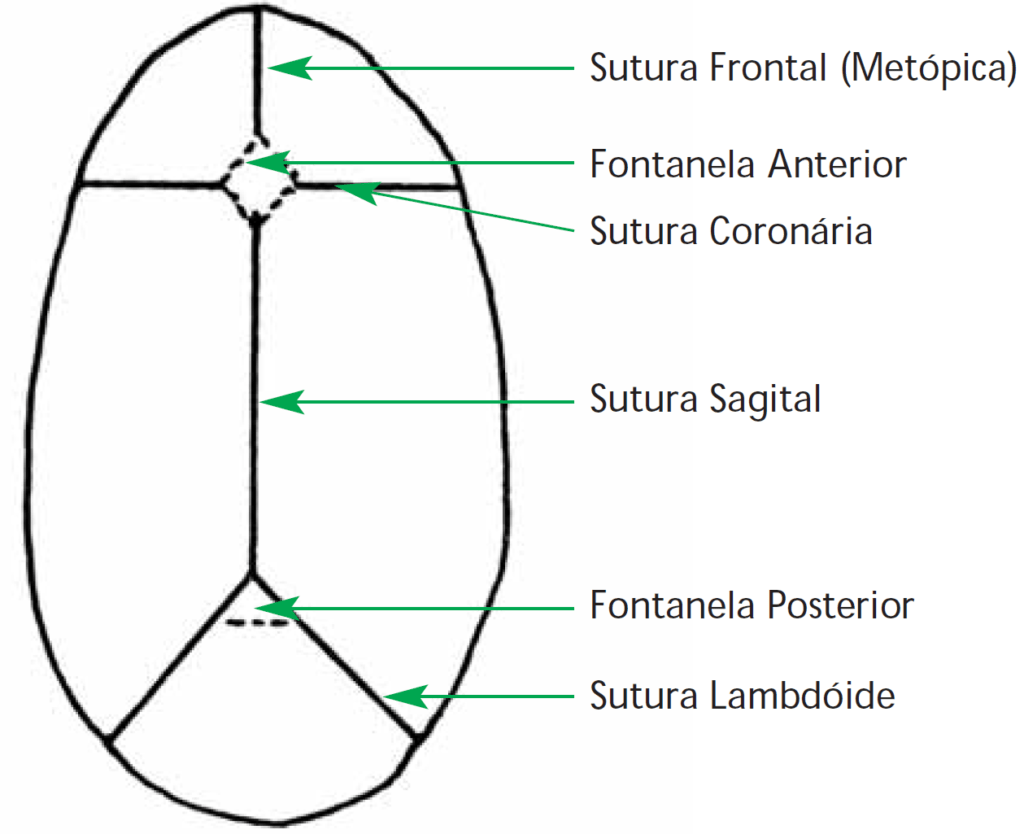
FIGURA 1-A. Crânio: fontanelas e suturas; visão esquemática superior abstraindo as observadas em visão lateral (Adaptado de SBP)
Fisiopatologia
A propósito da relação da estrutura óssea craniana com o conteúdo encefálico importa reter as seguintes noções básicas:
- o aumento de capacidade do crânio é um processo secundário ao aumento de volume das estruturas intracranianas;
- o tamanho da cabeça pode ser afectado pela espessura dos ossos do crânio;
- a dimensão e a forma do crânio ao longo do desenvolvimento resultam do equilíbrio entre esse estímulo e a capacidade de crescimento dos ossos ao longo das suturas;
- a ruptura de tal equilíbrio depende de três factores: moldagem interna, moldagem externa e encerramento precoce de uma ou várias suturas (craniossinostose).
Como exemplo de moldagem interna (situação em que o conteúdo craniano influencia a forma da cabeça) cita-se o aparecimento de bossas frontais observáveis nos casos de colecções extracerebrais benignas.
Como exemplos de moldagem externa (por forças externas ou pela acção do próprio peso) citam-se as deformações transitórias no pós-parto, nas primeiras semanas nos RN pré-termo e, as resultantes do decúbito dorsal ou lateral mantido, recomendado para dormir, na perspectiva da prevenção da morte súbita. (ver adiante)
Sobre o encerramento/ossificação precoce de suturas ou craniossinostose (com aspectos da patogénese ainda não totalmente esclarecidos), importa uma referência à chamada lei de Virchow: quando determinada sutura é precocemente encerrada, o crescimento ósseo é interrompido no sentido perpendicular à referida sutura; por compensação, o desenvolvimento do crânio ocorre no sentido paralelo, à custa das suturas não afectadas. O resultado final será o surgimento de deformações de grau variável e/ou assimetrias; por outro lado, a palpação da zona da sutura encerrada precocemente evidencia saliência óssea.
A repercussão clínica das craniossinostoses em termos de lesões do sistema nervoso é muito variável, dependendo da localização. Enquanto nalguns tipos poderá não se verificar qualquer lesão, noutros, por exemplo, poderá ser causa de atrofia óptica por alongamento do nervo óptico. (ver adiante)
1. MACROCEFALIA
Definição e semiologia
A macrocefalia, significando crânio de grandes dimensões, define-se pela verificação de perímetro cefálico acima do percentil 97 e crescimento excessivo da cabeça. Na prática, tal critério sobrepõe-se ao que é definido por alguns autores: perímetro cefálico > 2 desvios-padrão acima da média para uma determinada idade e sexo.
Como notas semiológicas importantes a propósito da definição (que tem limitações), importa salientar que:
- cerca de 2-3% da população considerada dentro da normalidade preenche os referidos critérios diagnósticos de macrocefalia;
- o perímetro cefálico pode ter uma velocidade de crescimento muito rápida traduzida pelo cruzamento de percentis (nomeadamente nos casos de lactentes em fase de recuperação de crescimento ou catch up growth), sobretudo se houver antecedentes de prematuridade ou de restrição de crescimento fetal;
- nem sempre um perímetro cefálico de valor elevado é sinónimo de crânio volumoso: por exemplo, em determinados casos de dolicocefalia o crânio terá um perímetro maior do que outro mais esférico, conquanto o volume de ambos seja o mesmo.
O Quadro 1 resume as principais causas de macrocefalia.
QUADRO 1 – Causas principais de macrocefalia
| isiológicas Lactente ex-pré-termo ou restrição de crescimento fetal, familiar constitucional, estatura elevada |
| Alterações predominantemente ósseas Raquitismo, hipofosfatasémia, acondroplasia, disostose crânio-facial de Crouzon, mucopolissacaridoses – gargoilismo ou doença de Hurler, etc. |
| Alterações da substância nervosa Megaencefalia, tumores cerebrais, neurofibromatose, gigantismo cerebral ou síndroma de Sotos |
| Alterações das meninges Derrame subdural, hematoma subdural |
| Alterações da circulação do líquido cefalorraquidiano Hidrocefalia congénita ou adquirida, ventriculite |
As situações de macrocefalia devem ser encaminhadas para centros especializados. (ver capítulo sobre alterações da migração neuronal)
2. MICROCEFALIA
Definição e classificação
A microcefalia ou diminuição do volume do crânio é definida pela verificação de perímetro cefálico abaixo do percentil 3 (ou inferior a 2 desvios – padrão/DP abaixo da média) em associação a velocidade lenta, anormal, do crescimento da cabeça.
Situações com DP entre < 2 e < 3 poderão comportar-se sob o ponto de vista neurológico como normais. Por outro lado, a incapacidade intelectual é praticamente uma constante nos casos de perímetro cefálico < 3 DP. Exceptuando nos casos de craniossinostose, a microcefalia implica sempre microencefalia, isto é, encéfalo anormalmente pequeno.
A microcefalia é classificada como primária quando resulta de aberração do desenvolvimento ou de agressão em fase precoce da neurogénese; a consequência é a diminuição do número ou das dimensões das células. São considerados diversos tipos de hereditariedade (AD, AR, ligada ao X), sendo mais favorável o prognóstico nas formas dominantes. Como regra, a microcefalia já é óbvia na data de nascimento.
A microcefalia é considerada secundária quando resulta de agressões ou noxas variadas actuando sobre um encéfalo previamente normal, ou no final do 3º trimestre da gravidez, ou durante o período perinatal (pós-natal precoce).
As situações de microcefalia devem ser encaminhadas para centros especializados.
O estudo imagiológico por TAC ou RM tem um papel fundamental na investigação etiopatogénica. Para além da possibilidade de visualização das estruturas ósseas, permite revelar eventuais calcificações, sugestivas de eventaul infecção pré-natal. (ver capítulo sobre RM e defeitos congénitos do SNC)
O Quadro 2 resume as principais causas de microcefalia.
QUADRO 2 – Causas principais de microcefalia
| Genéticas Familiar não associada a atraso do neurodesenvolvimento (microcefalia vera), forma autossómica recessiva associada a dificuldades de aprendizagem, associada a diversas síndromas (por ex. Menkes, Cornelia de Lange, Rubinstein – Taybi, Smith-Lemli-,Opitz, Seckel, etc. |
| Cromossómicas Associada a trissomias: 21-síndroma de Down, 18-síndroma de Edwards, 13-síndroma de Patau |
| Causas intrauterinas Infecções do grupo TORCHS, irradiação fetal, diabetes materna, fenilcetonúria ou aminoacidúria materna, etc. |
| Causas perinatais Sequelas de hipóxia-isquémia, de infecção do sistema nervoso central, de lesões traumáticas, de toxicidade bilirrubínica/kernicterus, etc.) |
3. CRANIOSSINOSTOSE
Aspectos epidemiológicos
A craniossinostose, quer na sua forma isolada, quer associada a outras anomalias congénitas integrando ou não síndromas, ocorre aproximadamente em 1 para 2.000 nados-vivos.
A situação mais frequente corresponde ao encerramento prematuro da sutura sagital, com um predomínio no sexo masculino de 3/1. A craniossinostose coronal surge com uma frequência aproximada de 20%, predominando no sexo feminino. As craniossinostoses metópica e lambdóide são mais raras.
A frequência com que se verifica associação de encerramento de duas ou mais suturas é cerca de 15%.
Estão descritas formas esporádicas e familiares, respectivamente com incidências de 1/1.700 a 2.500, e 1/25.000 nados-vivos.
Aspectos genéticos
Certas formas clínicas de craniossinostose são geneticamente determinadas; a este respeito, importa salientar 4 genes principais associados a formas sindrómicas de craniossinostose. Descrevem-se mais de 100 síndromas com craniossinostose associada.
A caracterização molecular dos genes associados a formas sindrómicas de craniossinostose é importante, pois permite fazer um diagnóstico mais preciso, especialmente durante o período neonatal, de forma a definir o tratamento e o seu possível resultado (como a eficácia duma intervenção cirúrgica craniana); igualmente, por permitir calcular o risco de recorrência na família.
O painel de testes inclui 10 mutações pontuais nos 4 genes principais associados a formas sindrómicas de craniossinostose: FGFR1 (Pfeiffer), FGFR2 (Apert, Crouzon e Jackson-Weiss), FGFR3 (Muenke e Seathre-Chotzen) e RAB23 (Carpenter). Com este painel de mutações é possível identificar a base molecular das formas mais frequentes e graves das síndromas genéticas de craniossinostose. De salientar que as mutações FGFR (gene do factor de crescimento dos fibroblastos) representam a maioria das formas sindrómicas.
Classificação
As craniossinostoses, quanto ao tipo, podem classificar-se em:
- simples, quando somente está afectada uma sutura;
- complexas, quando várias suturas estão afectadas;
- sindrómicas, quando existe associação a outros defeitos, constituindo síndromas genéticas perfeitamente definidas. (ver adiante)
Manifestações clínicas
Para além do sinal de alerta que é a dismorfia craniana, aponta-se a possibilidade de perda de visão. Pode observar-se também proptose na sinostose da sutura coronal e nas craniossinostoses complexas e sindrómicas.

FIGURA 2. Esquema de conformações anormais do crânio. A: Turricefalia; B: Braquicefalia; C: Escafocefalia; D: Plagiocefalia (Adaptado de SBP)
I – Craniossinostoses não sindrómicas
Seguidamente procede-se à descrição sucinta de diversas formas de craniossinostose, exemplificando-se esquematicamente com algumas destas na Figura 2 (designadas por A, B, C e D).
Escafocefalia ou craniossinostose longitudinal
ou sagital – C
Como se referiu antes, é a mais comum das craniossinostoses, predominando no sexo masculino. Resultando da ossificação precoce da sutura sagital, surge a forma de crânio em nave “com a quilha virada para cima”: crânio alongado no sentido ântero-posterior com proeminência dos ossos frontal e occipital; por conseguinte, verifica-se simultaneamente dolicocefalia. A fontanela anterior ou bregmática pode estar presente e desviada.
Dum modo geral não existe modificação importante do perímetro cefálico nem repercussão neurológica.
Plagiocefalia
Este termo significa genericamente obliquidade ou assimetria da forma da cabeça nos planos sagital ou coronal. Existem três formas de plagiocefalia: frontal, occipital e posicional.
Plagiocefalia frontal ou craniossinostose coronal unilateral/crânio oblíquo – D
Seguindo-se em frequência à escafocefalia, neste tipo verifica-se assimetria do crânio e face: depressão ou achatamento unilateral da fronte, elevação ipsilateral da órbita e sobrancelha e redução da cavidade orbitária com desvio do nariz para o lado não afectado. Tipicamente resulta da fusão prematura de uma sutura coronal e esfenofrontal. O índice cefálico pode estar normal, aumentado ou diminuído.
Plagiocefalia occipital lambdóide sinostótica
Resulta da fusão ou esclerose de uma sutura lambdóide, unilateralmente, com consequente achatamento occipital unilateral, procidência contralateral dos ossos frontal e occipital, o que determina forma trapezóide da cabeça. Esta discrania ocorre na proporção ~1/300.000 nados vivos. A forma bilateral é mais rara.
O diagnóstico diferencial da plagiocefalia sinostótica faz-se com a plagiocefalia posicional ou não sinostótica. (ver capítulo seguinte)
Trigonocefalia ou craniossinostose metópica ou frontal
A ossificação precoce da sutura frontal origina saliência ao longo do trajecto da mesma ou proeminência em forma de “quilha de barco”. Verifica-se hipotelorismo e a fontanela bregmática está sempre fechada. O crânio tem aspecto triangular com certa retracção das porções laterais das regiões frontais. As formas ligeiras são mais comuns.
Como a fusão ocorre em geral in utero, a criança evidencia esta anomalia ao nascer. De referir a probabilidade de associação a outras anomalias do encéfalo, tais como holoprosencefalia.
Turricefalia – A
A combinação mais frequente diz respeito à craniossinostose coronal e sagital. Tal pode conduzir a crescimento do crânio para cima em “torre” ou turricefalia; pode associar-se a braquicefalia (B).
Podendo estar associada a hipoplasia do maciço facial, esta modalidade conduz, em geral, a défice de crescimento do encéfalo e a insuficiência mental.
Braquicefalia (acro ou acrobraquicefalia) ou craniossinostose coronal bilateral – B
Nesta modalidade de craniossinostose verifica-se redução da distância ântero-posterior e aumento látero-lateral. A fronte apresenta-se achatada. A fontanela anterior, quando presente, está desviada para diante.
Pode associar-se turricefalia (A) e a sinais de hipertensão intracraniana.
Oxicefalia ou craniossinostose coronal e sagital
A manifestação mais típica da ossificação de todas as suturas é uma protuberância ou “gibosidade” na região da fontanela anterior originando a chamada oxicefalia.
Uma vez que se trata de deformação pouco notada, em geral o diagnóstico é tardio, quando surgem por vezes manifestações como convulsões ou sinais de hipertensão intracraniana. (Figura 3)
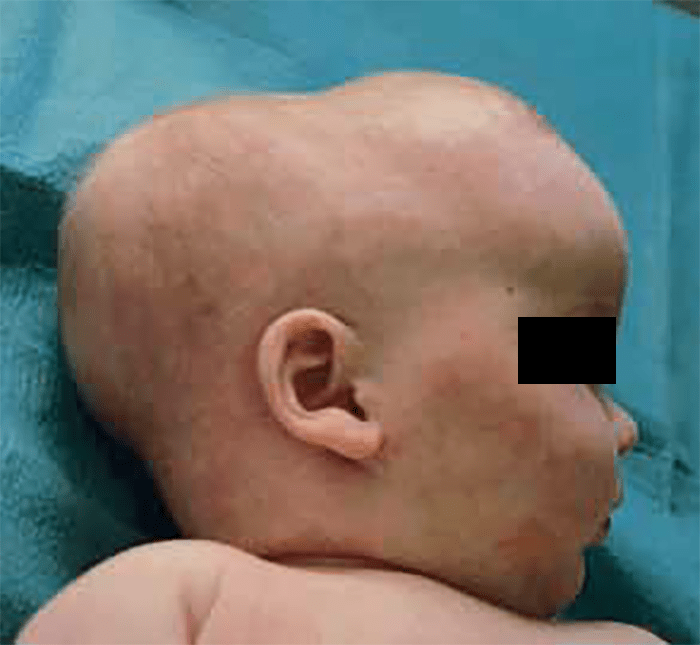
FIGURA 3. Aspecto de lactente com quadro de escafocefalia associada a oxicefalia. (NIHDE)
II – Craniossinostoses sindrómicas
No âmbito deste tipo de craniossinostoses, cabe salientar algumas das mais representativas entidades clínicas:
- síndroma de Crouzon com acantosis nigricans ou disostose craniofacial em que se verificam: exoftalmia, hipertelorismo, nariz curvo em “bico de papagaio” e prognatismo consecutivo a hipoplasia do maxilar superior. Trata-se de doença hereditária com transmissão autossómica dominante de expressividade variável (4p 16.3, FGFR3).
- síndroma de Apert ou acrocefalossindactilia de tipo I, em que as manifestações são similares às da síndroma de Crouzon, acrescentando-se sindactilia das mãos e pés. Esta afecção tem hereditariedade dominante, salientando-se que, na maioria das vezes, tem carácter esporádico relacionado com mutações de novo (10q26, FGFR2).
- síndroma de Carpenter ou acrocefalossindactilia do tipo II, em que existe dismorfismo facial, acrocefalia, braquiclinossindactilia das mãos e polissindactilia dos pés. A transmissão hereditária é de tipo autossómico recessivo com expressividade variável (16p12.1-q12, RAB23).
- síndroma de Pfeiffer ou acrocefalossindactilia do tipo V, caracterizada por turricefalia, anomalias faciais dismórficas, sindactilias discretas, polegares e dedos dos pés grandes. A transmissão hereditária é de tipo autossómico dominante com expressividade variável (8p11.2, FGFR1; 10q26, FGFR2).
Exames complementares
As craniossinostoses correspondem a situações diagnosticadas na sua maioria durante os períodos pré-natal (por ecografia) ou neonatal (por radiografia simples do crânio).
Havendo antecedentes familiares, e na perspectiva do diagnóstico sindrómico, poderá estar indicado o estudo do ADN, o qual pode ser obtido a partir das vilosidades coriónicas, líquido amniótico, sangue ou fibroblastos de biópsia fetal.
A radiografia simples do crânio é um exame considerado de eleição por confirmar a anomalia morfológica e permitir objectivar as suturas encerradas. (Figura 4)
A tomografia axial computadorizada (TAC) confirma o diagnóstico com mais rigor, permitindo estudo tridimensional. A ressonância magnética (RM) do encéfalo permite o diagnóstico de anomalias encefálicas associadas por vezes.
O diagnóstico diferencial deve fazer-se com as microcefalias ligadas a compromisso encefálico primitivo em que o atraso do neurodesenvolvimento não é acompanhado de encerramento precoce das suturas nem de hipertensão intracraniana.
Tratamento e prognóstico
O tratamento das craniossinostoses tem os seguintes obectivos: 1- assegurar o crescimento normal do cérebro; 2- prevenir a hipertensão intracraniana; 3- prevenir as sequelas neurológicas, oculares e auditivas; 4- contribuir para a melhoria estética do crânio e face.
Nos casos de escafocefalia a actuação é apenas estética. Como regra geral, pode estabelecer-se que, quando está indicado tratamento, este é sempre cirúrgico, da competência dos especialistas de neurocirurgia pediátrica; se a intervenção for realizada nos primeiros seis meses de vida serão obtidos melhores resultados.
O prognóstico depende de vários factores: tipo de craniossinostose, idade do diagnóstico, existência ou não de anomalias congénitas associadas, de hipertensão intracraniana, estrabismo, atrofia óptica, e de neurodesenvolvimento afectado.
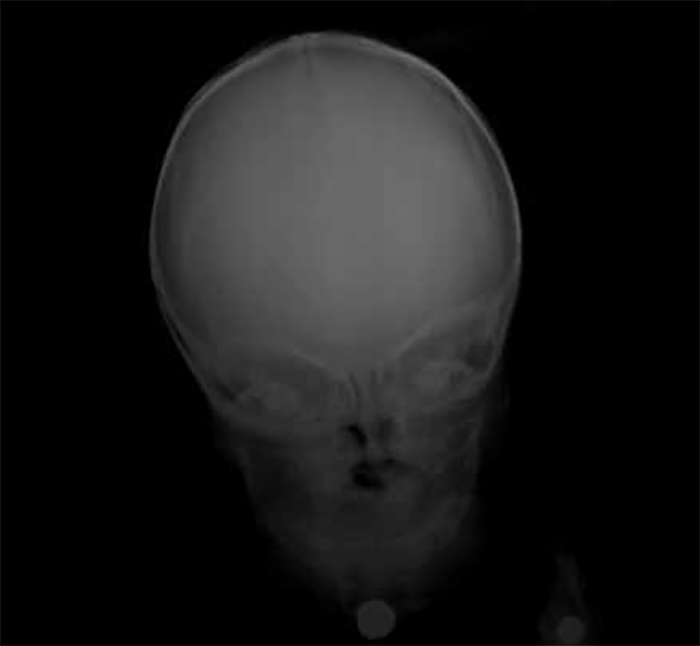
FIGURA 4. Aspecto radiográfico do crânio em lactente com turricefalia, não sendo visíveis as suturas por sinostose. (NIHDE)
BIBLIOGRAFIA
Aicardi J. Diseases of the Nervous Central System. London: Mac Keith Press, 2009
Campagnoni AT, el al (eds). Developmental Neuroscience. Basel: Karger, 2008
Cheek WR. Atlas of Pediatric Neurosurgery. Philadelphia: Saunders, 1996
Galdo A, Cruz M. Exploracion Clínica em Pediatria. Barcelona: Editorial Jims, 2000
Gill D, O´Brien N. Paediatric Clinical Examination. Edinburgh: Churchill Livingstone, 2003
www.infocefalia.com (Acesso em Janeiro de 2019)
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Loman WS, Flannery AB. Evidence-based care of the child with deformational plagiocephaly. Part I: assessment and diagnosis. J Pediatr Health Care 2012; 26: 242-250
Losee JE, Mason AC. Deformational plagiocephaly: Diagnosis, prevention and treatment. Clin Plast Surg 2005; 32: 53-64
McLone DG. Pediatric Neurosurgery. Philadelphia: Saunders, 2001
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Nield LS, Brunner MD, Kamat D. The infant with a misshapen head. Clin Pediatr (Phila) 2007; 46: 292-298
Rasmussen SA, Yazdy MM, Frias JL, Honein MA. Priorities for public health research on craniosynostosis. Am J Med Genet 2008; 146 A: 149-158
Ridgway EB, Weiner HL. Skull deformities. Pediatr Clin North Am 2004; 359-387
Roach ES (ed). Pediatric Neurology. Philadelphia: Elsevier, 2019
Speltz ML, Kapp-Simon KA, Cunningham M, et al. Single-suture craniosynostosis: a review of neurobehavioral research and theory. J Pediatr Psychol 2004; 29: 651-668
Swaiman KF, Ashwal S, Ferriero DM, Schor NF. Swaiman’s Pediatric Neurology. Principles and Practice. Philadelphia: Elsevier Saunders, 2012
Zitelli BJ, McIntire SC, Norwalk AJ (eds). Zitelli and Davis’ Atlas of Pediatric Physical Diagnosis. Philadelphia: Elsevier, 2012