Sistematização
Como foi referido no capítulo dedicado aos defeitos do tubo neural, as anomalias congénitas do SNC são detectadas no RN com uma frequência de 0,5 a 5%.
Neste capítulo são abordados:
- os defeitos da migração, proliferação e diferenciação neuronais;
- as anomalias da segmentação e da divisão cerebrais;
- e, sucintamente, a porencefalia.
Trata-se de situações raras, por vezes com diversidade de anomalias associadas, e com consequências diversas: o espectro de manifestações varia entre casos detectados sem repercussões clínicas relevantes (heterotopia mínima de neurónios), e outros com repercussões devastadoras incluindo incapacidade intelectual e motora graves, e síndromas acompanhadas de convulsões.
1. DEFEITOS DA MIGRAÇÃO, PROLIFERAÇÃO e DIFERENCIAÇÃO NEURONAIS
No que respeita a alterações da migração neuronal, investigações experimentais recentes chamaram a atenção para o papel de determinados genes e suas mutações.
Nesta alínea é feita uma abordagem sucinta de algumas formas clínicas em que a imagiologia, designadamente a RM, tem um papel fundamental na respectiva identificação.
Lisencefalia
Também designada agiria, esta anomalia rara é caracterizada por ausência de circunvoluções cerebrais/cérebro “liso”, com fissura sílvica vestigial; o aspecto macroscópico do cérebro é o de cérebro fetal com cerca de 12-16 semanas de gestação. (Figura 1A: imagem de RM – CE)
Lisencefalia do tipo I
A este grupo pertence à síndroma de Miller-Dieker caracterizada por fenótipo especial: fronte estreita e saliente, hipertelorismo, nariz curto com anteversão das narinas, orelhas de implantação anómala e micrognatismo, microcefalia, convulsões, atraso do desenvolvimento, hipocrescimento, hipoplasia do nervo óptico e microftalmia.
Esta forma clínica depende de expressão defeituosa do gene LIS1(17p13.3) e do gene da lisencefalia (LIS-1).
Nos casos em que se comprova associação a disgenesia do corpo caloso e a hipoplasia do cerebelo e do tronco cerebral foram identificadas mutações no gene TUBA1A (12q22-q24).
Lisencefalia do tipo II
O protótipo desta forma clínica é a síndroma de Walker-Warburg, caracterizada por hidrocefalia, agíria, defeito do cerebelo, displasia da retina, onfalocele e distrofia muscular congénita, hipotonia e morete precoce.
A transmissão hereditária é do tipo autossómico recessivo, tendo-se identificado vários genes responsáveis, tais como POMT1, POMT2, ISPD, FKTN, etc., situados respectivamente nos cromossomas 9q14, 14q24.3, 7p21, 9q31-33.
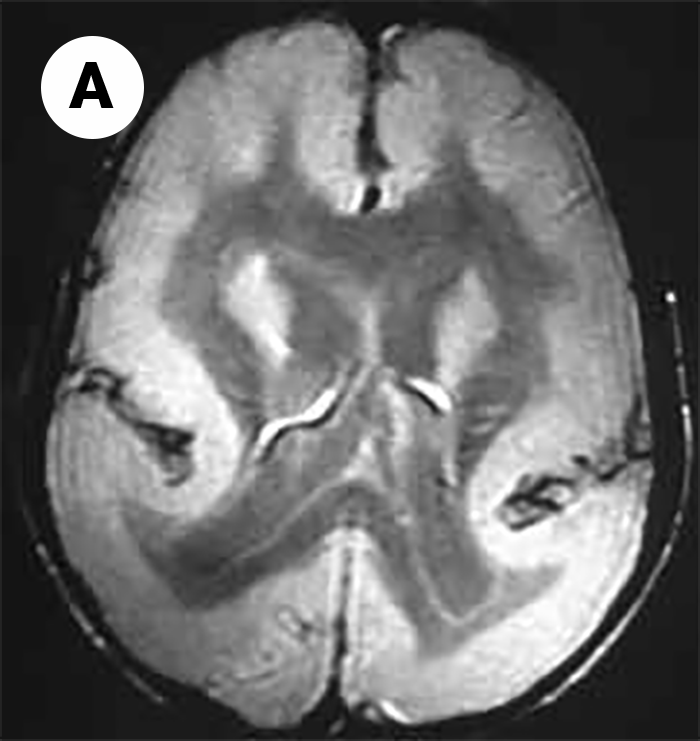
FIGURA 1A: Imagem de lisencefalia. (RM-CE)
Esquizencefalia
Nesta anomalia verifica-se a presença de fendas unilaterais ou bilaterais ou ao nível dos hemisférios cerebrais. Nalguns casos os achados da RM permitem visualizar fenda de comunicação entre o ventrículo e o espaço craniano extra-axial; muitas destas fendas/”comunicações” estão tapetadas por substância cinzenta anormal. Como manifestações clínicas refere-se atraso mental, convulsões refractárias, microcefalia, e tetraparésia espástica quando as fendas são bilaterais. Se a fenda for unilateral pode verificar-se hemiparésia. (Figura 1B: imagem de RM – CE)
Esta anomalia pode ser determinada geneticamente (gene EMX2, 10q26.1).
Descrevem-se dois tipos I e II, conforme respectivamente os bordos estajam abertos ou fechados.
Agenesia do corpo caloso
A esta anomalia, clinicamente muito heterogénea (desde formas assintomáticas e QI normal, até síndromas neurológicas complexas acompanhadas de défice mental), está associada hereditariedade ligada ao X, ou autossómica dominante; pode igualmente estar ligada a anomalias cromossómicas (trissomias 8 e 18) e associada a certas doenças hereditárias do metabolismo.
Na síndroma de Shapiro a agenesia do corpo caloso associa-se a episódios recidivantes de hipotermia e diaforese.
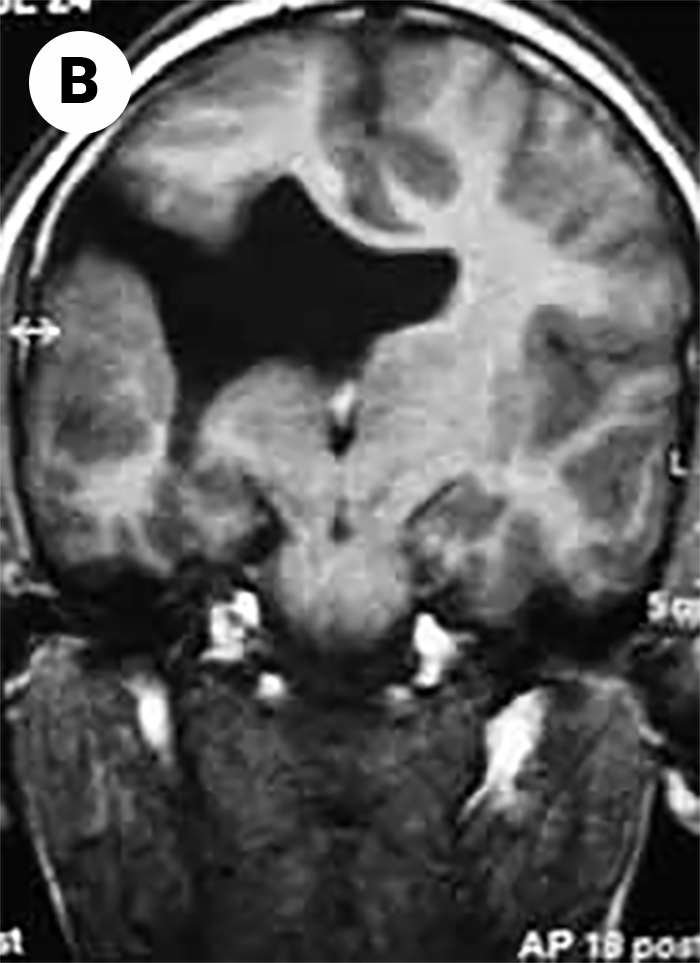
FIGURA 1B: imagem de esquizencefalia. (RM-CE)
A síndroma de Aicardi (quadro complexo caracterizado essencialmente por atraso mental, espasmos em flexão/hipsarritmia, convulsões refractárias, coriorretinite “em queijo Gruyères” e anomalias vertebrais/hemivértebras) está tipicamente também associada a agenesia do corpo caloso. Predominando no sexo feminino, admite-se anomalia do cromossoma X, a que corresponde elevada letalidade no sexo masculino.
Na síndroma de Anderman (gene SLC12A6) existe associação a neuropatia periférica.
Agenesia dos nervos cranianos
Esta anomalia, por vezes associada a diversas situações clínicas, compreende ausência de certos nervos cranianos ou dos respectivos núcleos originando sinais clínicos diversos, por ex. ptose palpebral congénita, fenómeno de Marcus Gunn (concomitância de movimentos de sucção e pestanejo/sincinésia, etc.). Na síndroma de Moebius verifica-se paralisia facial bilateral.
Heterotopias neuronais
Neste defeito existem colecções de neurónios em localiazação anómala.
Descrevem-se três grupos:
Heterotopias nodulares subependimárias periventriculares
Este grupo tem na sua base diversas alterações genéticas tais como, entre outras: ligadas ao cromossoma X(Xq28, gene da filaminaA, FLNA) e outras dependentes de gene autossómico (20q13.13), gene ARFGEF2).
Heterotopias subcorticais e marginais glioneurais
Este grupo está relacionado com alterações nos genes LIS1 (17p13.3) e DCX (Xq22.3-q23).
Heterotopias laminares subcorticais
Este grupo está limitado praticamente ao sexo feminino, com transmissão dominante ligada ao cromossoma X(Xq22.3-q23, gene DCX). No sexo masculino esta alteração genética pode associar-se a lisencefalia.
Megalencefalia, macrocefalia e hemimegalencefalia
A megalencefalia define-se como desenvolvimento precoce de cérebro anormalmente grande. Existe uma forma familiar benigna (com hereditariedade autossómica dominante, sobretudo no sexo masculino. Os ventrículos cerebrais podem ser de dimensões normais ou moderadamente de grandes dimensões.
Na macrocrânia ou macrocefalia observa-se crânio grande, nem sempre acompanhado de cérebro grande (constitucional ou familiar, igualmente observado nas síndromas de Sotos, de Weaver, acondroplasias, etc.).
A hemimegaloencefalia define-se como hipertrofia difusa cerebral unilateral, secundária a anomalias na proliferação e migração neuronais (gene L1-CAM, Xq28), a qual se manifesta por macrocefalia nem sempre assimétrica, atraso psicomotor e epilepsia precoce rebelde ao tratamento.
Microcefalia
Define-se pela verificação de perímetro cefálico inferior a 2 DP do considerado normal para a idade, sexo e idade gestacional. Consideram-se dois grupos: microcefalia primária e microcefalia secundária.
Uma forma extrema de microcefalia primária é o chamado microcérebro radial a que corresponde cérebro com < 50 g.
Num grupo característico de microcefalias primárias familiares, de hereditariedade autossómica recessiva, estão implicados fundamentalmente sete genes, desde MCPH1, 8p23; MCPH2, 19q13.12; MCPH3, 9q33, etc., a MCPH7, 1p32.3-p33. Estas situações, em geral no contexto de consanguinidade, estão associadas a fenótipo em que ressaltam, entre outras, as seguintes características: nariz proeminente, orelhas desproporcionadamente grandes, micrognatismo, e incapacidade intelectual.
2. ANOMALIAS DA SEGMENTAÇÃO E DIVISÃO CELULARES
Holoprosencefalia
Trata-se duma anomalia resultante de clivagem defeituosa do prosencéfalo com incidência da ordem de 1/5.000-1/16.000 e susceptível de ser diagnosticada no período pré-natal a partir da 10ª semana. Compreende três formas: alobar, semilobar, e lobar.
De transmissão autossómica recessiva, por vezes associada a diabetes materna, em cerca de 50% dos casos, existe associação com trissomias 13, 15 e 18; Estão implicados diferentes genes, tais como: SHH (7q36), ZIC2 (13q32), SIX3 (2p21), TGIF (18p11.3), etc..
Como manifestações clínicas de anomalias da segmentação e divisão cerebrais, são notórias as anomalias faciais: fenda palatina, lábio leporino, ciclopia, cebocefalia, incisivo central único, e agenesia pré-maxilar. Através da RM, a forma lobar evidencia ausência de separação dos hemisférios e ventrículos laterais, substituídos por ventrículo único central.
Anomalias do septum pellucidum
Este tipo de anomalias pode ter duas expressões:
- quisto do septum pellucidum que pressupõe a persistência duma cavidade intrasseptal própria do feto (cavum) para além dos 4-5 meses de vida, a qual é raramente sintomática;
- ausência do septum pellucidum a qual provoca a fusão dos ventrículos laterais para originar uma cavidade única central. Este defeito pode estar associado a hipoplasia dos nervos ópticos, malformações do prosencéfalo e disfunção hipotalâmica (displasia septo-óptica ou síndroma de Morsier com anomalias nos genes HESX1/PAX3, 3p21.1-p21.2, e SOX2, 3q27), que se manifesta por nistagmo, ambliopia, hipopituitarismo e défice motor. O diagnóstico é confirmado através de exames de imagem –TAC e RM.
3. PORENCEFALIA
A designação genérica de porencefalia refere-se às situações em que existem quistos ou cavidades intracerebrais. Para além da etiopatogénese relacionada com defeito do desenvolvimento (congénita), tal quadro morfológico pode também ser adquirido na sequência de enfarte tecidual.
Esta anomalia, por vezes associada a outras (encefalocele, microcefalia, etc.), manifesta-se fundamentalmente por insuficiência intelectual, hemi ou tetraparésia espástica, atrofia óptica e convulsões.
Nota final: Como foi referido no capítulo 196, os defeitos do cerebelo, as hidrocefalias, as cranossinostoses e síndroma de Klippel-Feil são abordadas noutros capítulos, de modo integrado.
AGRADECIMENTO
O autor agradece à Dra. Eulália Calado a cedência das imagens. (Figura 1)
BIBLIOGRAFIA
Aicardi J. Diseases of the Nervous Central System. London: Mac Keith Press, 2009
Barkovich AJ, Kuzniecky RI, Jackson MD, e tal. Classification system for malformations of cortical development. Neurology 2001; 57: 2168-2178
Campagnoni AT, el al (eds). Developmental Neuroscience. Basel: Karger, 2008
Campagnoni AT. Developmental Neuroscience. Basel: Karger, 2008
Cheek WR. Atlas of Pediatric Neurosurgery. Philadelphia: Saunders, 1996
Gill D, O´Brien N. Paediatric Clinical Examination. Edinburgh: Churchill Livingstone, 2003
Clark GD. The classification of cortical dysplasias through molecular genetics. Brain Dev 2004; 26: 351-362
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Rasmussen SA, Yazdy MM, Frias JL, Honein MA. Priorities for Public Health Research on Craniosynostosis. Am J Med Genet 2008; 146 A: 149-158
Roach ES (ed). Pediatric Neurology. Philadelphia: Elsevier, 2019
Roberts B. Neuronal migration disorders. Radiol Technol 2018; 89: 279-295
Speltz ML, Kapp-Simon KA, Cunningham M, et al. Single-suture craniosynostosis: a review of neurobehavioral research and theory. J Pediatr Psychol 2004; 29: 651-668
Swaiman KF, Ashwal S, Ferriero DM, Schor NF. Swaiman’s Pediatric Neurology. Principles and Practice. Philadelphia: Elsevier Saunders, 2012
Sztriha L. Spectrum of corpus callosum agenesis. Pediatr Neurol 2005; 32: 94-10