Conceitos fundamentais
Etimologicamente e tal como foi escrito pela primeira vez por Nicolas Andry em meados do século XVIII, no título de um tratado que escreveu sobre a correcção das deformidades na criança, Ortopedia significa: “arte de corrigir, nas crianças, as deformidades do corpo”. Rapidamente, porém, estendeu-se este termo ao estudo e tratamento de outros tipos de patologia com repercussão dominante sobre o esqueleto, sem distinção da idade do doente, e generalizou-se a partir do final do século XIX a toda a patologia do aparelho locomotor. Na actualidade, embora a denominação deste ramo da Medicina se tenha mantido inalterada, traduz um âmbito muito mais amplo, podendo-se definir, de forma abreviada, e de acordo com a Academia Americana de Cirurgia Ortopédica, como “a especialidade que compreende o diagnóstico, tratamento, reabilitação e prevenção das lesões e doenças do sistema músculo-esquelético”.
Este sistema músculo-esquelético desempenha basicamente três tipos de funções primordiais: a de depósito e reserva de elementos indispensáveis à vida do organismo, de suporte e protecção de estruturas nobres importantes, e a de protagonista na postura e na mobilidade dos diferentes segmentos anatómicos em que habitualmente dividimos o corpo humano.
Em virtude desta sua última função, cumpre um papel fundamental na locomoção e, através dela, na vida de relação do indivíduo de qualquer idade, contribuindo na criança, de forma secundária mas determinante, para o seu desenvolvimento.
Devido às particularidades anátomo-fisiológicas do esqueleto da criança e adolescentes que os tornam distintos do adulto, adquire nessas idades o estatuto de sector especializado – a Ortopedia Pediátrica – com diferenciação e individualidade próprias dentro da actividade ortopédica geral, constituindo o objectivo temático da presente Parte deste Tratado.
Entre as referidas particularidades, a mais importante e típica destas idades será o crescimento, como capacidade intrínseca e única do esqueleto infantil, desde a origem, para aumentar as suas dimensões e modelar a sua morfologia até assumir por completo, no final da adolescência, as características do esqueleto do adulto.
Este crescimento, apesar das variações de velocidade com que se efectua, dependentes, entre outras circunstâncias, da idade cronológica da criança e da região anatómica considerada, realiza-se de forma permanente, desde o aparecimento do esboço ósteo-articular primitivo, cerca da oitava semana da gestação, até à completa maturidade esquelética; por isso, o crescimento representa um factor condicionante básico de toda a patologia ortopédica na idade pediátrica.
Assim, face a determinada situação patológica ortopédica, tratando-se de um organismo em crescimento e desenvolvimento, às alterações primárias decorrentes da própria lesão músculo-esquelética, há que associar sempre uma avaliação do potencial de crescimento remanescente no segmento anatómico afectado, porque dele depende a magnitude e o tipo das alterações secundárias resultantes da referida lesão. Este potencial tanto pode actuar em desfavor como em benefício do doente, o que significa a possibilidade de originar e agravar deformidades, ou de as prevenir e corrigir se, pelo contrário, for bem aproveitado e conduzido.
A ambivalência deste factor dá relevância a uma outra característica importante e bem visível da Ortopedia Pediátrica, na sua vertente terapêutica; referimo-nos à prevenção das alterações ou deformidades do crescimento. Como é evidente, esta prevenção (que, em determinadas circunstâncias poderá ser possível na fase pré-natal), será sempre muito mais fácil de executar e com resultados mais gratificantes do que a correcção das deformidades, uma vez estabelecidas.
Ainda no que se refere ao tratamento destas situações, seja ele preventivo ou curativo, será conveniente salientar a necessidade de o equacionar sempre também numa perspectiva de longo prazo, para a eventualidade de futuras exigências terapêuticas adicionais na idade adulta que podem ficar comprometidas se ignorarmos ou minimizarmos esta condição.
Na presente Parte optou-se por uma abordagem sucinta das situações mais comuns que afectam o sistema músculo-esquelético, dividindo-a em dois sectores. No primeiro faremos algumas considerações sumárias sobre factores de ordem geral, relativos às características do esqueleto da criança e à sintomatologia habitual da sua patologia, para, no segundo sector, subdividido em duas secções, nos referirmos à patologia não traumática e traumática deste grupo etário. Na impossibilidade evidente de nos alongarmos nestas considerações, tivémos como objectivos essenciais transmitir os aspectos práticos que permitam chegar ao diagnóstico correcto da situação e a uma avaliação das suas consequências, proporcionar a informação básica relativa à terapêutica adequada, e prover orientações para o encaminhamento atempado dos doentes para o especialista.
Considerações gerais sobre semiologia
O esqueleto na idade pediátrica distingue-se do esqueleto do adulto, conforme já dissémos, por apresentar um determinado número de características que o individualizam no seu comportamento não só fisiológico, habitual, como também na forma como responde às agressões que o atingem. As mais importantes são: a sua maior elasticidade, uma grande capacidade de remodelação, a presença de um periósteo espesso e a existência das cartilagens de crescimento, nomeadamente das chamadas cartilagens fisárias, de conjugação ou simplesmente fises.
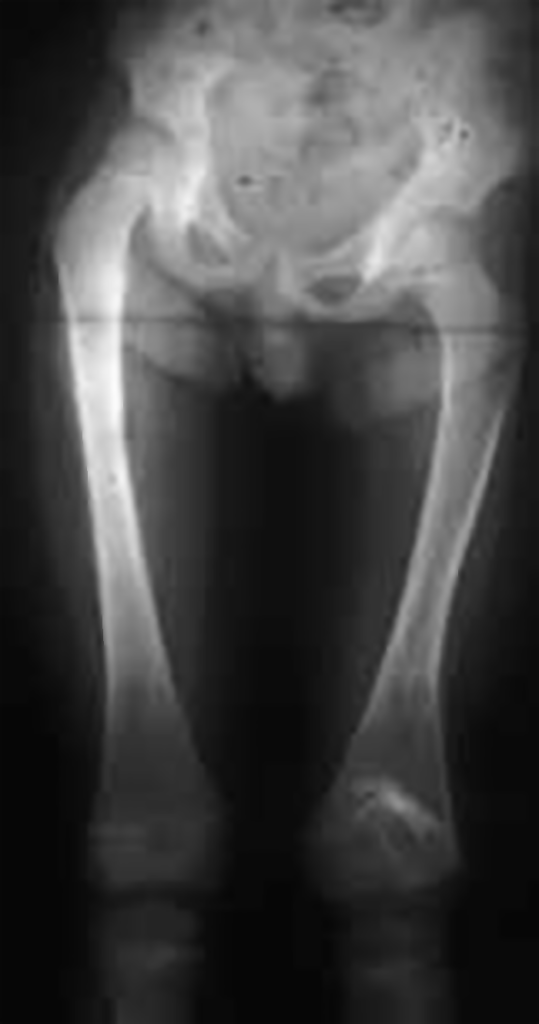
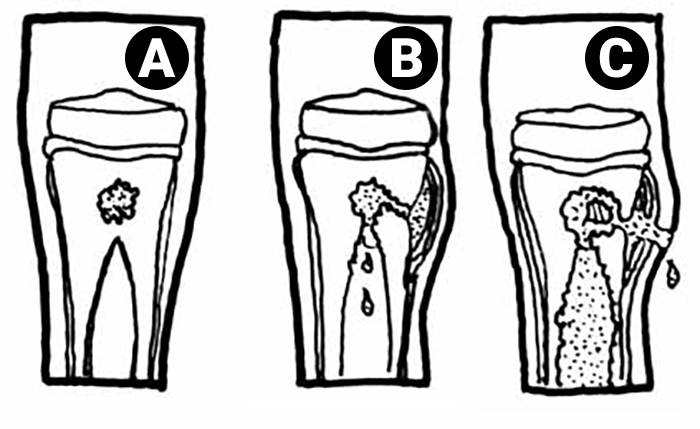
Se as primeiras lhe permitem ter uma maior plasticidade, adaptando-se melhor às forças a que se encontra sujeito, mantendo ou readquirindo a configuração que lhe é própria, a espessura do periósteo, além de constituir um invólucro robusto do tecido ósseo subjacente, contribui, pela sua grande capacidade de regeneração óssea, juntamente com as cartilagens de crescimento, para o aumento gradual das dimensões e forma da peça óssea correspondente. Nos ossos longos, protagonistas do crescimento do esqueleto na sua dimensão mais aparente, ou seja, em comprimento, entre as distintas cartilagens de crescimento existentes, as mais importantes do ponto de vista fisiopatológico e clínico, são as chamadas cartilagens de conjugação. Trata-se de estruturas com alto nível de diferenciação histológica e funcional, responsáveis pelo crescimento longitudinal do osso, localizadas nas respectivas metáfises e interpostas no tecido ósseo; aquelas constituem, pela sua natureza cartilagínea, um segmento de menor resistência mecânica, susceptível de se fracturar ou descolar do tecido ósseo subjacente, dando assim origem a transtornos do crescimento, de gravidade variável. Se, como dissémos, as podemos considerar estruturas frágeis do ponto de vista mecânico, por outro lado, ao estarem interpostas no tecido ósseo desta zona anatómica dos ossos longos, servem habitualmente de barreira à expansão de lesões de natureza infecciosa ou tumoral localizadas na sua vizinhança.
As referidas características do esqueleto infantil, cujo potencial é máximo à nascença, vão-se atenuando gradualmente com a idade, acabando por desaparecer no final da adolescência, quando o indivíduo atinge a idade adulta.
Passando agora à abordagem dos aspectos semiológicos revelados por um exame clínico adequado, importa considerar que, se a qualidade deste exame é fundamental na prática clínica de qualquer especialidade, na Ortopedia tem uma importância capital porque, com frequência, dá-nos de imediato o diagnóstico, ou para ele nos orienta, conseguindo-se chegar a uma conclusão acertada mediante o recurso a poucos e simples exames complementares. Na Ortopedia Pediátrica, embora esta característica se mantenha, ao abranger doentes numa faixa etária em que se produzem profundas transformações orgânicas e comportamentais, esse exame assume aspectos particulares e impõe condicionalismos a que devemos atender.
Assim, na observação clínica de uma criança pequena, quase sempre irrequieta ou amedrontada, não é possível aplicar a metodologia habitual, longa e exaustiva, utilizada no exame do adulto.
Requerem-se por isso, da parte do médico, imaginação e experiência para saber seleccionar rapidamente, a partir da história, o conjunto de manobras exploratórias, de execução simples e breve, adequadas à detecção da causa das queixas actuais.
Por outro lado, é preciso saber que a sintomatologia de determinadas situações patológicas nem sempre se revela de forma constante no decurso das várias etapas do crescimento; por exemplo, as infecções dos discos intervertebrais (discites) na criança pequena podem manifestar-se inicialmente por uma incapacidade para a marcha; mais tarde, apenas por alterações do estado geral; e, na adolescência, por dor local na coluna vertebral. Portanto, o exame clínico deverá ser realizado tendo sempre em conta, não só a suspeita da patologia em causa, como também a idade do doente.
Reportando-nos agora apenas aos sinais e sintomas locais das afecções ortopédicas, e abstraindo dos sintomas gerais eventualmente associados, são três os achados semiológicos principais, básicos, habitualmente presentes neste tipo de situações: dor, deformidade e impotência funcional. É porém evidente que em Pediatria, a comprovação exacta do predomínio relativo de qualquer um deles será muito variável, dependendo não só do tipo da doença e sua fase de evolução, como também até da capacidade de avaliação e comunicação das queixas por parte do próprio doente, o que estará em relação directa com a idade e respectivo desenvolvimento neuropsíquico. Será, por isso, muito diferente a maneira como uma criança pequena, outra em idade escolar, e um adolescente, manifestam a presença e intensidade de sintomas, competindo ao médico a responsabilidade e o engenho necessários à sua adequada caracterização.
A dor é um sintoma importante a valorizar devidamente porque em Ortopedia Pediátrica e, contrariamente ao que se passa no adulto, tem uma causa orgânica em mais de 80% dos casos. Porém, na criança pequena, incapaz de se exprimir de forma adequada, este sintoma manifesta-se muitas vezes apenas pelo choro mais ou menos contínuo ou intenso, aumentando com a tentativa de mobilização da parte afectada, e na recusa em mobilizar ou utilizar o segmento anatómico do membro atingido pela lesão. Na criança mais velha e no adolescente, já será possível e obrigatório investigar a localização exacta da dor e sua eventual irradiação.
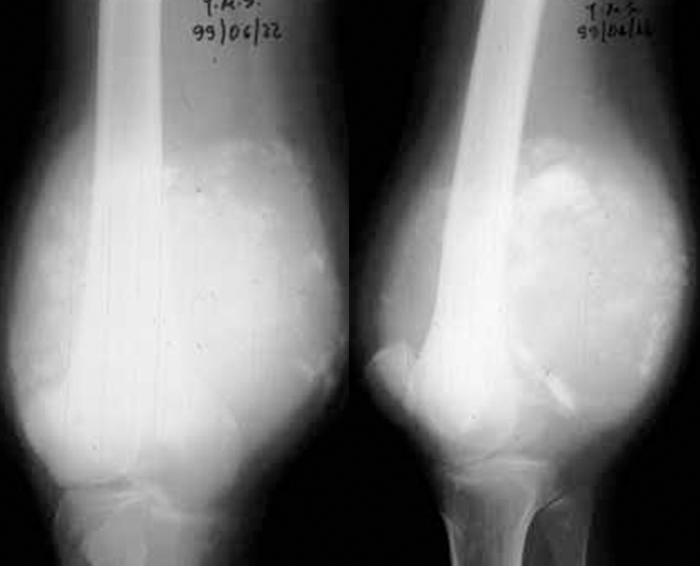
A sua duração e natureza, bem como as circunstâncias que determinaram o seu aparecimento e as que a aliviam ou a agravam, interferindo ou não com o sono (esclarecendo-nos sobre a sua intensidade e permanência), são outros tantos aspectos que interessa averiguar, por serem orientadores de um possível diagnóstico. Assim, por exemplo, uma dor persistente, sem causa aparente, não muito intensa, localizada na zona metafisária e prolongando-se ao longo de semanas, pode ser sugestiva de lesão tumoral. A dor aguda, intensa, pulsátil, justa-epifisária ou articular faz suspeitar de lesão inflamatória ou infecciosa.
Quanto à deformidade, tomada no sentido lato de alteração da forma ou da aparência habitual, tanto dos membros como do tronco (ou de qualquer um dos seus segmentos anatómicos), é necessário sublinhar que a criança se adapta facilmente à mesma, contrariamente ao que se passa no adulto, não lhe causando em geral grande limitação funcional.
Os pais são os primeiros a detectá-la e a fornecerem toda a informação desejada, mas é importante caracterizar bem o que, neste âmbito, se entende por patológico, principalmente no caso de este tipo de queixas surgir isolado. Com grande frequência os pais recorrem ao médico porque descobriram no filho o que, no seu entender, consideram ser deformidades, esperando daquele a confirmação da anomalia e exigindo a imediata actuação terapêutica. Acontece, porém, que na maioria dos casos, os invocados problemas não passam de situações sem qualquer significado patológico, traduzindo variantes da normalidade que se corrigem muitas vezes espontaneamente durante o desenvolvimento subsequente da criança.
Haverá, por isso, que investigar e interpretar correctamente certas alterações, enquadrando-as no conjunto da restante sintomatologia, e inquirindo especialmente sobre a sua localização, natureza e duração (quando e como começou; se tem sido progressiva, e rapidez dessa progressão). A impotência ou incapacidade funcional é outro dos sintomas dominantes na patologia ortopédica; trata-se do achado relacionado com a dor e a deformidade, e no grupo etário mais baixo pode constituir o único aparente. É mais evidente quando se estabelece de forma aguda e relacionado com um episódio recente, por exemplo, de natureza traumática ou inflamatória. Outras vezes instala-se de forma insidiosa e gradual, tornando-se menos perceptível, devido aos mecanismos de compensação funcional que a criança facilmente encontra e aos quais se adapta. Todos estes aspectos deverão, por isso, ser convenientemente investigados e valorizados na colheita da história clínica.
Os três sintomas/sinais principais que acabámos de mencionar estão sempre relacionados entre si, podendo haver predomínio de um ou outro consoante a patologia presente e as circunstâncias próprias do doente. No entanto, como princípio orientador, será importante não esquecer o conhecido aforismo: “a criança que se queixa tem quase sempre razão até prova em contrário”.
Isto significa que, nestas idades, a grande maioria dos doentes com sintomatologia evidente do tipo referido, tem uma causa orgânica subjacente que a explica e que importa investigar. No adolescente, tal não é tão evidente, aproximando-se do que se passa nos adultos: muitas vezes as queixas deste foro podem ter um componente emocional ou psíquico que as potencia ou determina, com o que será necessário contar para uma interpretação correcta das mesmas.
Considerando-se os sintomas/sinais descritos como os mais frequentes e notórios colhidos na anamnese e exame objectivo de doentes desta faixa etária com patologia ortopédica, torna-se depois necessário proceder à sua caracterização e interpretação com rigor.
Como na maioria das situações ortopédicas – incluindo muitas das doenças sistémicas com repercussão músculo-esquelética – as lesões têm carácter predominantemente regional ou local; é, por isso, conveniente adoptar uma estratégia definida na execução do referido exame objectivo. Embora cada observador possa seguir a que mais lhe convier, julgamos aconselhável sistematizar essa abordagem clínica, sugerindo, por exemplo, começar-se pela observação global do doente, passando depois à observação regional (membros e tronco), particularmente na zona das queixas actuais, para terminar na área anatómica exacta das referidas queixas.
O pormenor com que se procederá a estas distintas etapas dependerá do tipo de patologia e das circunstâncias particulares do doente, aspectos estes primariamente esclarecidos pela anamnese, com a qual, porém, todos os achados do exame objectivo devem ser permanentemente confrontados e testados, como única forma de se chegar a uma interpretação fidedigna de toda a informação recolhida e, daí, a um diagnóstico correcto.
Sublinha-se que um exame objectivo adequado exige a colaboração do doente e familiares, o que neste grupo etário, principalmente na criança pequena, não é fácil de conseguir. Irá depender da sua idade, desenvolvimento, comportamento, educação e relacionamento com os familiares directos (que, por vezes dificultam mais do que ajudam) e da paciência, capacidade de improvisação e experiência do médico que o atende. Sem estes atributos onde prevalece o chamado bom senso clínico, arriscamo-nos a transformar este exame numa cena de luta inglória com a criança e os familiares, com total impossibilidade de se chegar a conclusões válidas.
Por isso, a estratégia acima referida, é apresentada apenas como orientação genérica, devendo ser adaptada a cada caso concreto, segundo as suas características peculiares, não hesitando o clínico em passar imediatamente à última fase, a do exame local se for julgado conveniente, em vez de esgotar a débil capacidade de atenção e colaboração da criança em manobras exploratórias sem utilidade prática imediata na elaboração do diagnóstico.
Nunca será demais repetir que o exame objectivo deve ser realizado nas melhores condições possíveis no que diz respeito às instalações, com o doente despido e na presença dos pais ou familiares. A sua execução, independentemente da citada estratégia, custuma obedecer a uma sistematização que compreende as três etapas clássicas de inspecção, palpação e mobilização. Se é preferível e didacticamente mais correcto que estas etapas se processem de acordo com a referida ordem, nada impede que a experiência do clínico e o tipo de patologia presente determinem a modificação do esquema indicado, permitindo realizá-las em simultâneo.
Na Ortopedia Pediátrica, tal como acontece noutra áreas da Medicina, para a confirmação do diagnóstico não são necessários habitualmente múltiplos e dispendiosos exames complementares; na maioria das vezes os referidos exames complementares de diagnóstico resumem-se aos exames laboratoriais clássicos para avaliação global da situação, a que se acrescentam os específicos em função da área da patologia infecciosa e inflamatória, e aos exames imagiológicos; salientam-se a radiologia simples do esqueleto, a tomografia axial computadorizada (TAC), a ressonância magnética (RM), a ecografia e a cintigrafia. Com este leque de possibilidades no âmbito referido é quase sempre possível confirmar ou excluir o diagnóstico clínico sem que se tenha de recorrer a provas mais complicadas e caras.
Sistematização
Nesta Parte serão abordadas de forma necessariamente sucinta e genérica, algumas das situações mais correntes do foro ortopédico que afluem às consultas de Pediatria e para as quais os ortopedistas são muitas vezes solicitados a dar parecer. Procuraremos dar uma noção da sua frequência, importância do ponto de vista clínico e orientação terapêutica. O Quadro 1 sistematiza os tópicos a abordar nos capítulos seguintes.