Definição e importância do problema
Os hemangiomas são tumores benignos do endotélio vascular resultantes de um processo de vasculogénese, no qual os precursores das células endoteliais dão origem a novos vasos.
Por motivos de ordem estética, tal patologia tem certo impacte psicossocial na criança e famílias, implicando por parte do clínico e/ ou profissional de saúde esclarecimento sobre a história natural da mesma.
Aspectos epidemiológicos
Os hemangiomas infantis são os tumores vasculares mais frequentes na criança, com uma incidência de 2,6 a 4,5% nos lactentes. Os factores de risco associados ao desenvolvimento de hemangiomas são a prematuridade, raça caucasiana, sexo feminino (1,4-3:1) e baixo peso de nascimento. Alguns estudos identificaram outros factores tais como: idade materna avançada, pré-eclâmpsia, placenta prévia e gestações múltiplas.
Etiopatogénese
A etiopatogénese desta situação não está totalmente esclarecida. Têm sido propostas várias teorias para explicar as alterações no decurso da vasculogénese antes mencionada conduzindo ao desenvolvimento deste tipo de tumores: proliferação vascular induzida por hipóxia de tecidos, mutação somática das células endoteliais progenitoras possivelmente associada ao aumento da produção de mediadores que estimulam a proliferação daquelas, ou embolização de células placentárias.
História natural
Os hemangiomas infantis têm uma evolução típica que os distingue das malformações vasculares. Surgem nos primeiros dias a semanas de vida e podem ser precedidos nalguns casos por uma lesão macular ou telangiectasia. Apresentam uma fase de proliferação rápida (fase proliferativa) nos primeiros meses de vida, seguida de uma fase de estabilização (período temporário de equilíbrio entre a proliferação de células endoteliais e a sua apoptose) que pode durar até ao final do primeiro ano de vida. É pouco frequente ocorrer proliferação depois deste período.
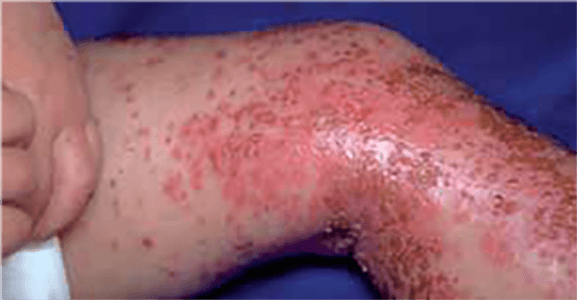
Depois da fase proliferativa e da de estabilização, segue-se uma fase de regressão gradual espontânea, em que o componente vascular é substituído por um tecido fibroadiposo. Esta fase de regressão tem duração variável, podendo prolongar-se até aos 9 anos. Estima-se que em cerca de metade dos hemangiomas, a involução do hemangioma está associada a lesões cutâneas residuais, como telangiectasia, pele redundante, cicatrizes ou tecido fibroadiposo.
Diagnóstico diferencial
O diagnóstico diferencial destes tumores inclui: – as malformações vasculares (que estão presentes ao nascer e não proliferam nem regridem; – outros tumores vasculares (granuloma piogénico, hemangioendotelioma kaposiforme); e – os hemangiomas congénitos ( também presentes ao nascer e que podem ser “rapidamente involutivos”, ou não involutivos).
A expressão da isoforma 1 do transportador de glicose (GLUT1) é exclusiva dos hemangiomas infantis.
Classificação
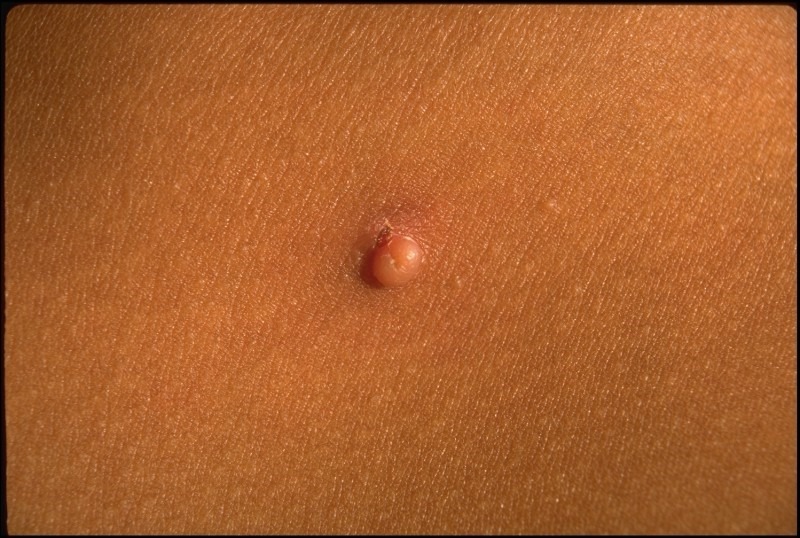
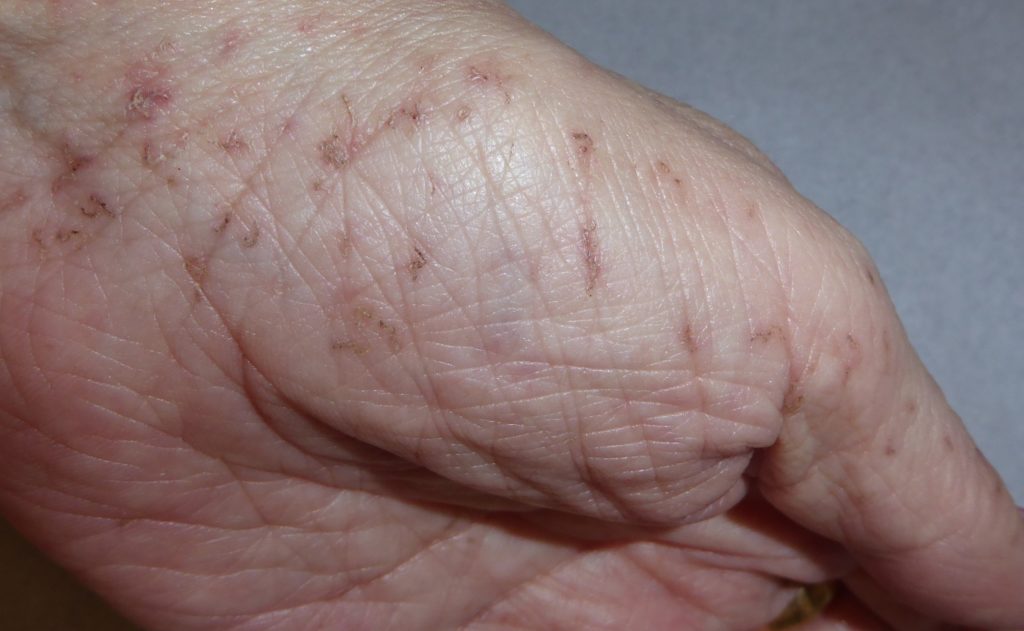
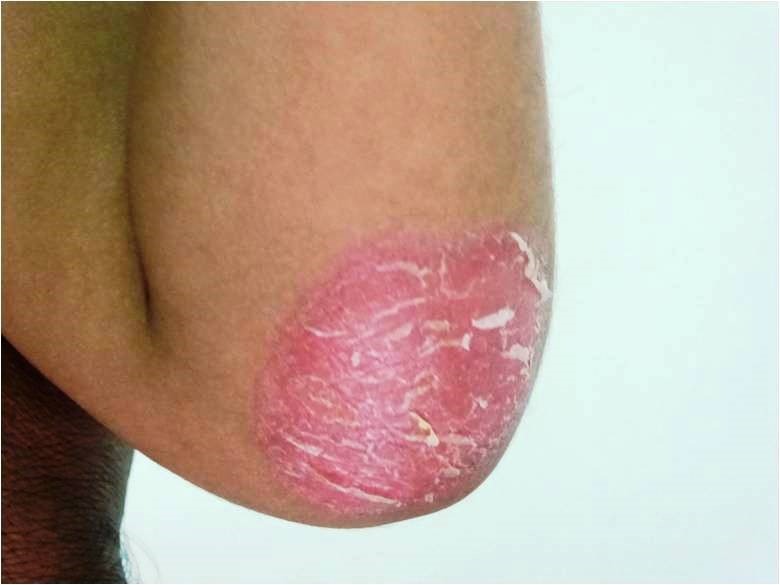
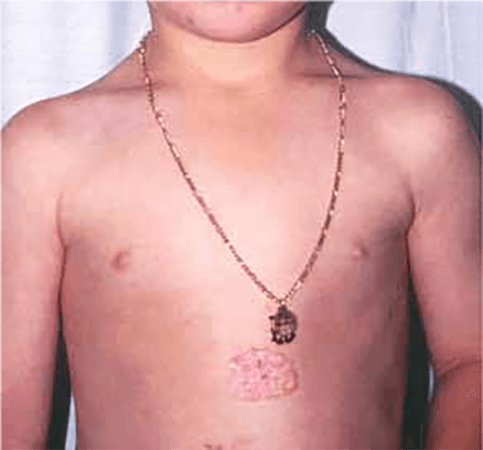
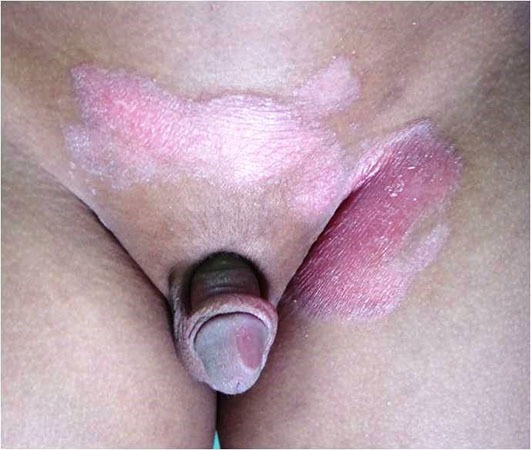
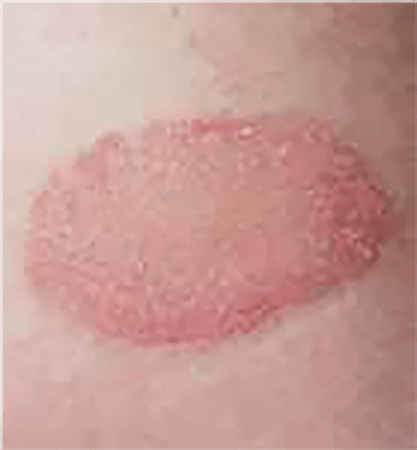
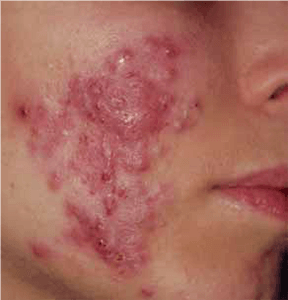
Os hemangiomas podem ser superficiais, profundos ou mistos. (Figura 1)
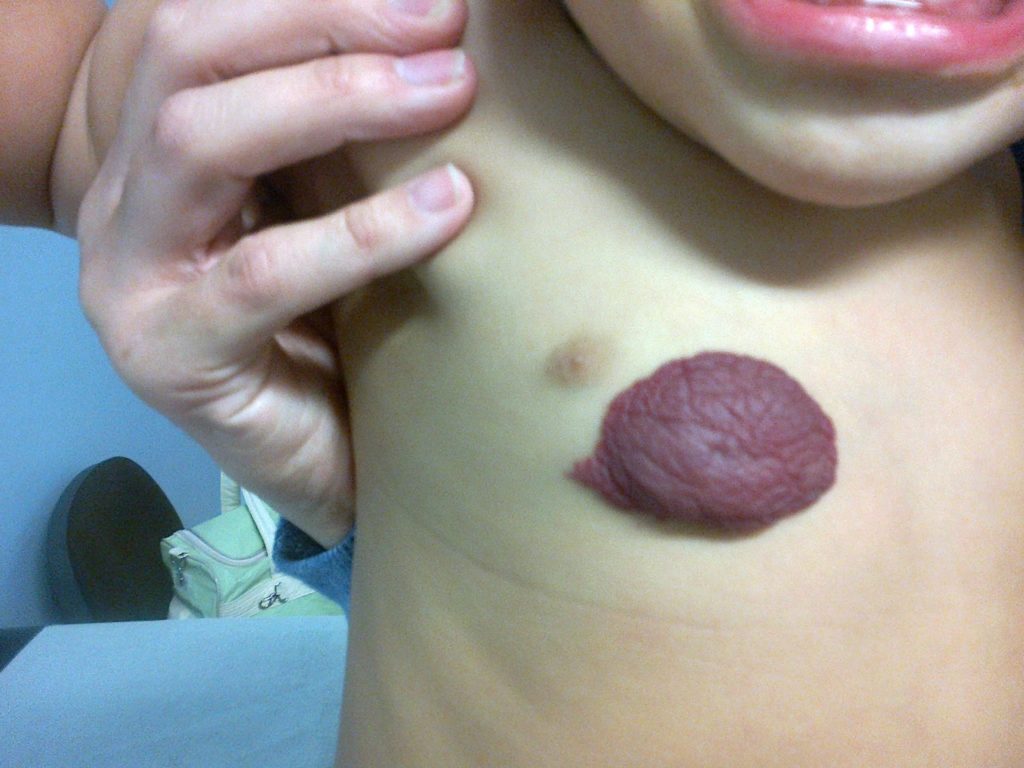
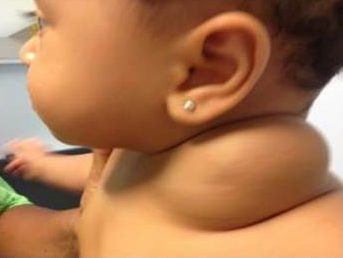
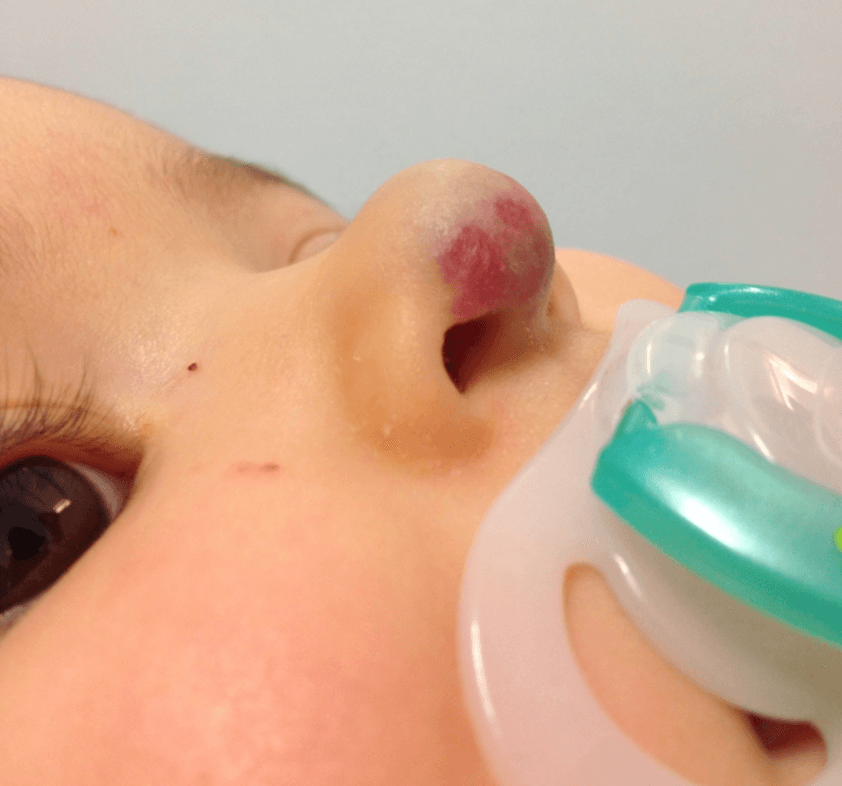
FIGURA 1. A) hemangioma superficial; B) hemangioma profundo; C) hemangioma misto
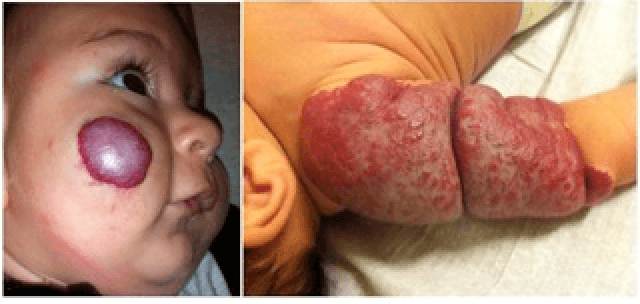
FIGURA 2. A) hemangioma focal; B) hemangioma segmentar
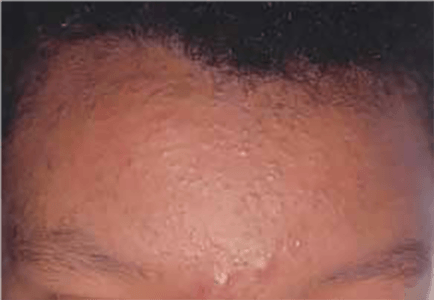
Os hemangiomas superficiais são os mais comuns; apresentam uma coloração avermelhada, não se distinguindo componente subcutânea.
Os hemangiomas profundos podem estar associados a uma coloração azulada da pele (historicamente conhecidos como “cavernosos”).
Os hemangiomas superficiais crescem em média até aos 5 meses de vida, enquanto os profundos podem apresentar uma fase proliferativa até aos 12 a 14 meses de idade.
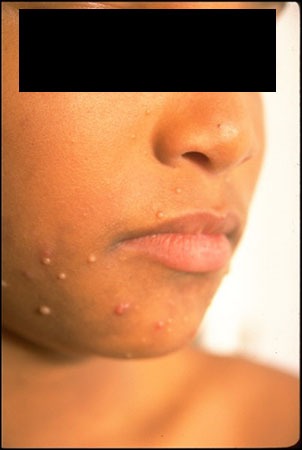
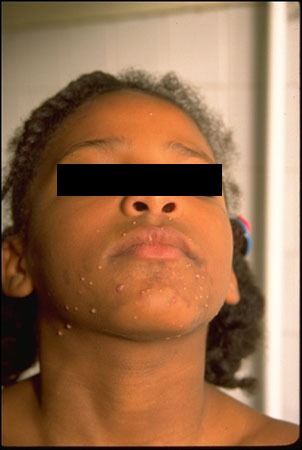
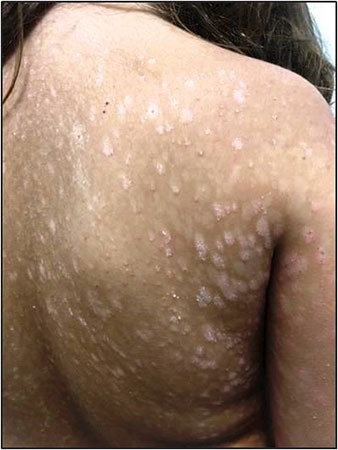
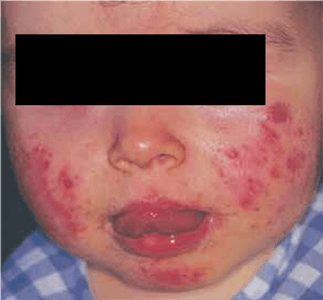
Com base na sua configuração anatómica, os hemangiomas podem ainda ser classificados em focais ou segmentares (Figura 2). Os hemangiomas focais são lesões bem circunscritas com relevo, enquanto os segmentares são lesões achatadas em placa com uma distribuição geográfica num segmento.
Manifestações clínicas
A cabeça e o pescoço são as regiões mais frequentemente envolvidas (60%), seguindo-se o tronco (25%) e as extremidades (15%). Na sua maioria, os hemangiomas são únicos, mas em 20% dos lactentes afectados as lesões são múltiplas. A presença de cinco ou mais hemangiomas pode estar associada a hemangiomas viscerais, nomeadamente hepáticos. Os hemangiomas cervicais ou mandibulares com distribuição “em barba” podem estar associados a uma lesão concomitante da via aérea, evidenciando frequentemente estridor.
Complicações
Apesar de na maioria dos casos os hemangiomas infantis serem tumores inócuos que regridem ao longo do tempo, alguns podem complicar-se devido às suas características ou localização,. As complicações, mais frequentes no sexo feminino, ocorrem em cerca de 10% dos casos, hemangiomas infantis. Numa perspectiva prática e didáctica, consideram-se quatro grupos:
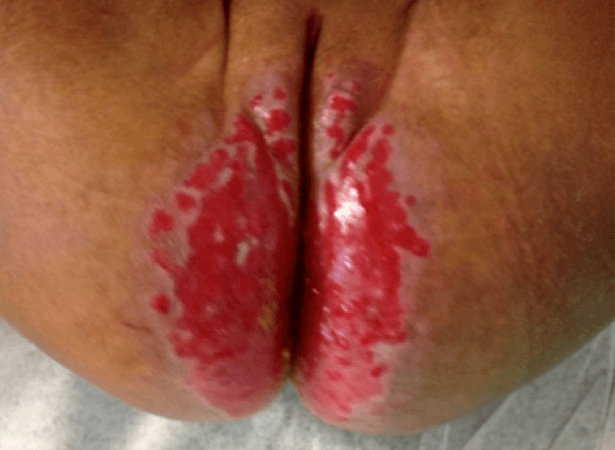
- Ulceração (a complicação mais frequente) – ocorre principalmente nos hemangiomas de grandes dimensões, do períneo ou localizados nas superfícies mucosas, zonas intertriginosas ou zonas de fricção.
- Desfiguração permanente – ocorre principalmente nos hemangiomas de grandes dimensões, nos segmentares, pedunculados ou da face, principalmente do lábio, nariz e pavilhão auricular.
- Compromisso de função – hemangioma perioral, que pode comprometer a amamentação; hemangioma periorbitário que, dependendo da localização e dimensões, pode condicionar exoftalmia, ambliopia, astigmatismo, estrabismo ou obstrução do canal lacrimal.
- Risco de vida – hemangiomas da via aérea, condicionando obstrução; hemangiomas hepáticos múltiplos; ou hemangiomas de grandes dimensões (ditos na gíria médica “gigantes”) podem causar insuficiência cardíaca de alto débito.
Síndromas e associações
Nalguns doentes, os hemangiomas infantis podem surgir associados a anomalias congénitas. Um hemangioma grande e segmentar da face, couro cabeludo e/ou pescoço pode estar incluído na associação PHACE (malformações da fossa Posterior, Hemangioma, malformações Arteriais/Aórticas, Cardíacas, e do olho / Eye). Esta associação pode incluir ainda defeitos da rafe esternal ou supraumbilical.
Por outro lado, no caso de hemangioma da linha média lombo-sagrada ou do períneo, é importante excluir a síndroma PELVIS/SACRAL/LUMBAR – diferentes acrónimos usados para caracterizar a associação a defeitos estruturais dos genitais externos, urológicas, anorrectais e mielopatia (disrafismo, lipomielomeningocele).
Exames complementares
O diagnóstico de hemangioma infantil é essencialmente clínico, sendo os métodos de imagem reservados para casos de lesão profunda sem componente cutâneo associado e para estudo de co-morbilidades associadas. (Quadro 1)
QUADRO 1 – Hemangiomas infantis e exames complementares com indicações específicas
| Hemangioma periorbitário | Avaliação oftalmológica para exclusão de alterações visuais, ponderar RM órbita |
| Hemangioma mandibular e sinais de dificuldade respiratória, nomeadamente estridor | Avaliação por ORL para exclusão de hemangioma da via aérea |
| ≥ 5 hemangiomas | Ecografia abdominal para exclusão de hemangioma hepático |
| Grande hemangioma segmentar da face ou couro cabeludo | Excluir síndroma PHACE: RM e angio-RM da cabeça e pescoço, ecocardiograma, avaliação por oftalmologista |
| Hemangioma da linha média lombo-sagrada | RM da coluna para exclusão de disrafismo espinhal |
Tratamento
Uma vez que na sua maioria todos os hemangiomas regridem espontaneamente, a terapêutica, não habitualmente necessária, está reservada para os hemangiomas complicados ou com risco de complicação.
Existem várias modalidades terapêuticas:
a) Terapêutica sistémica
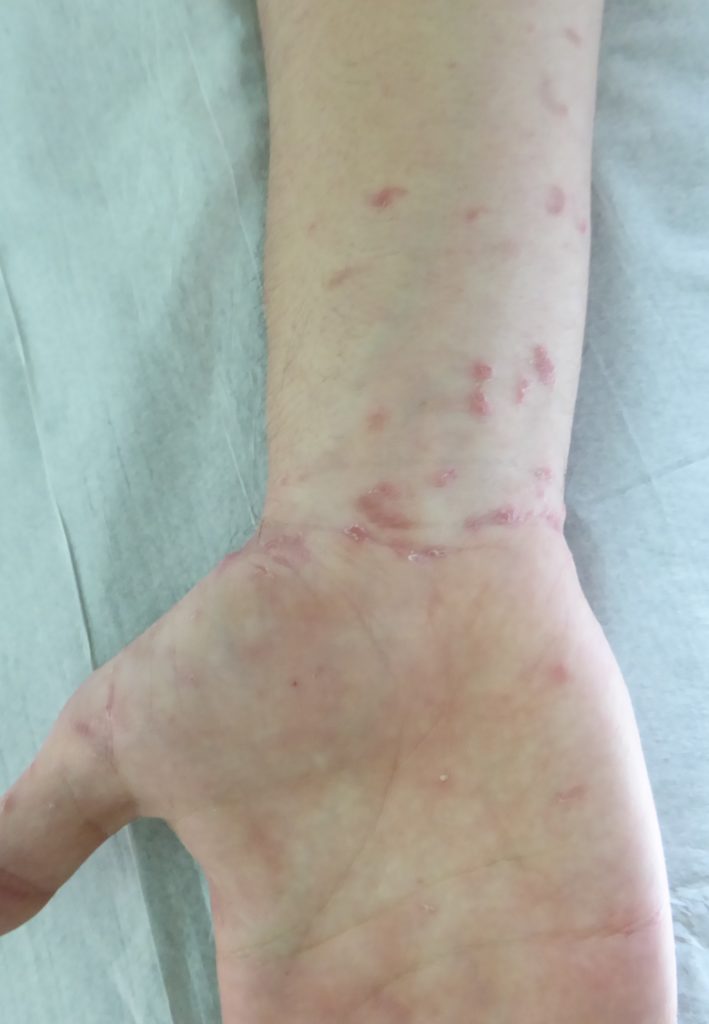
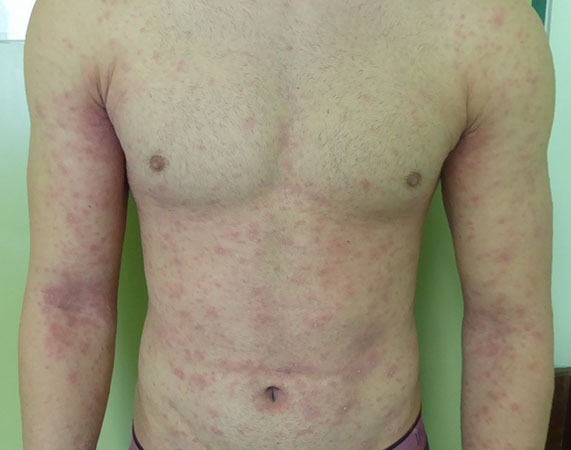
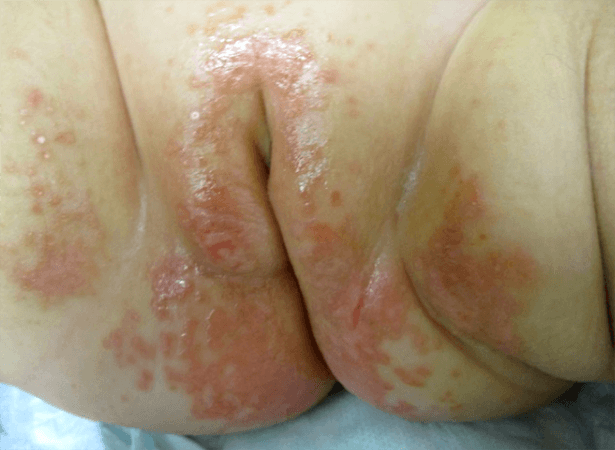
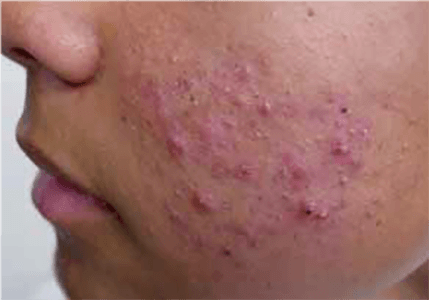
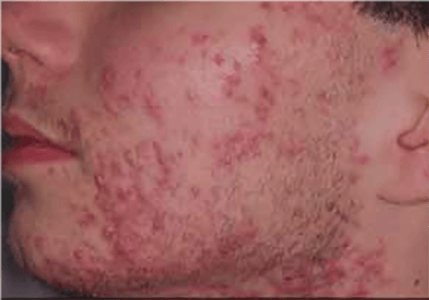
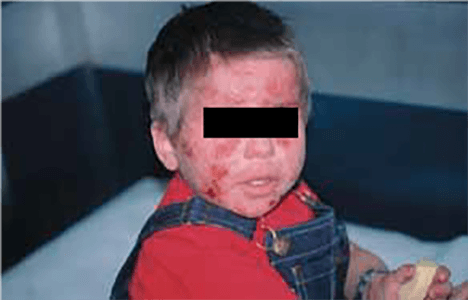
O propranolol (beta-bloqueante não selectivo) é actualmente a primeira opção com bons resultados na indução da involução destes tumores. (Figura 3)
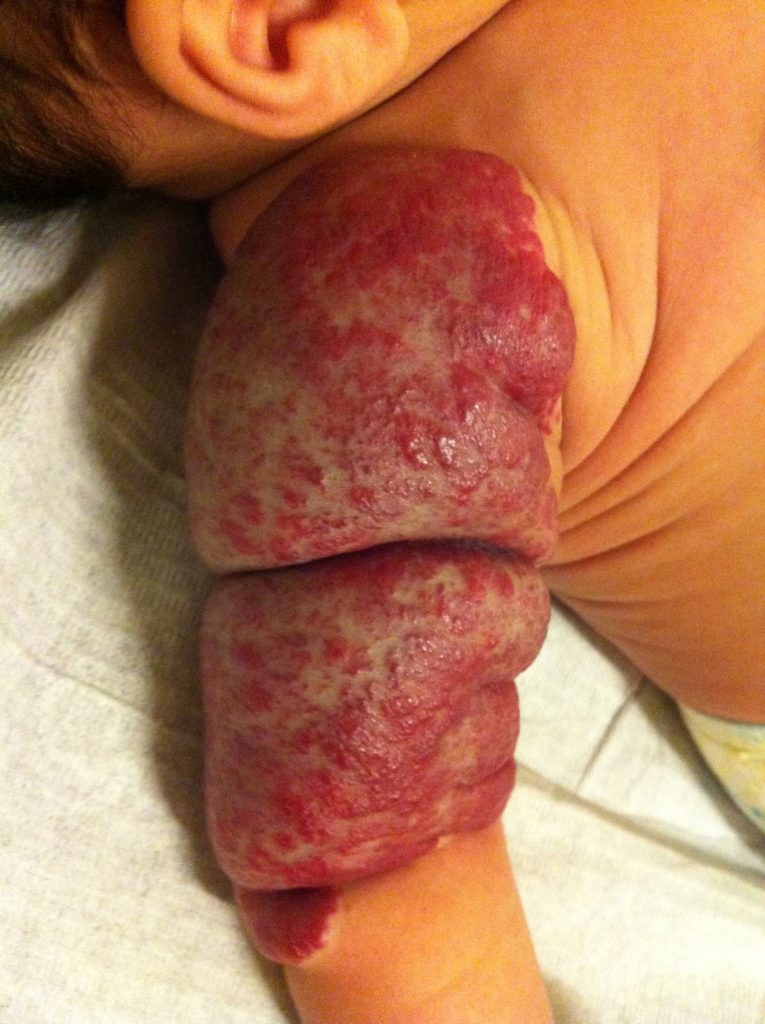
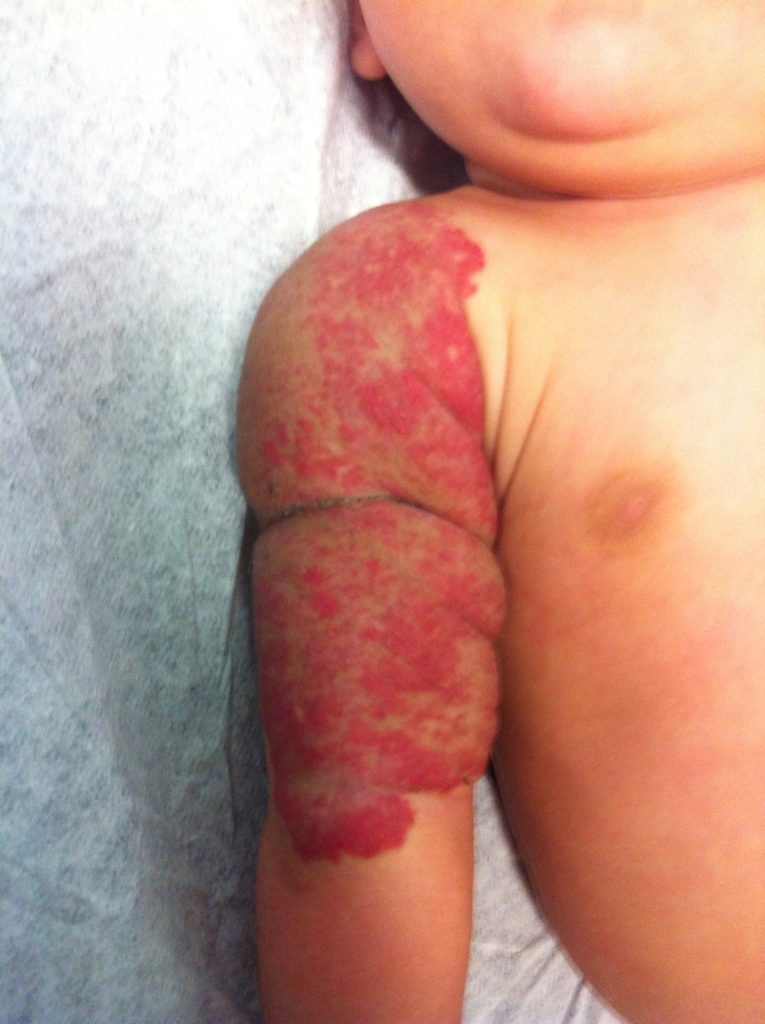
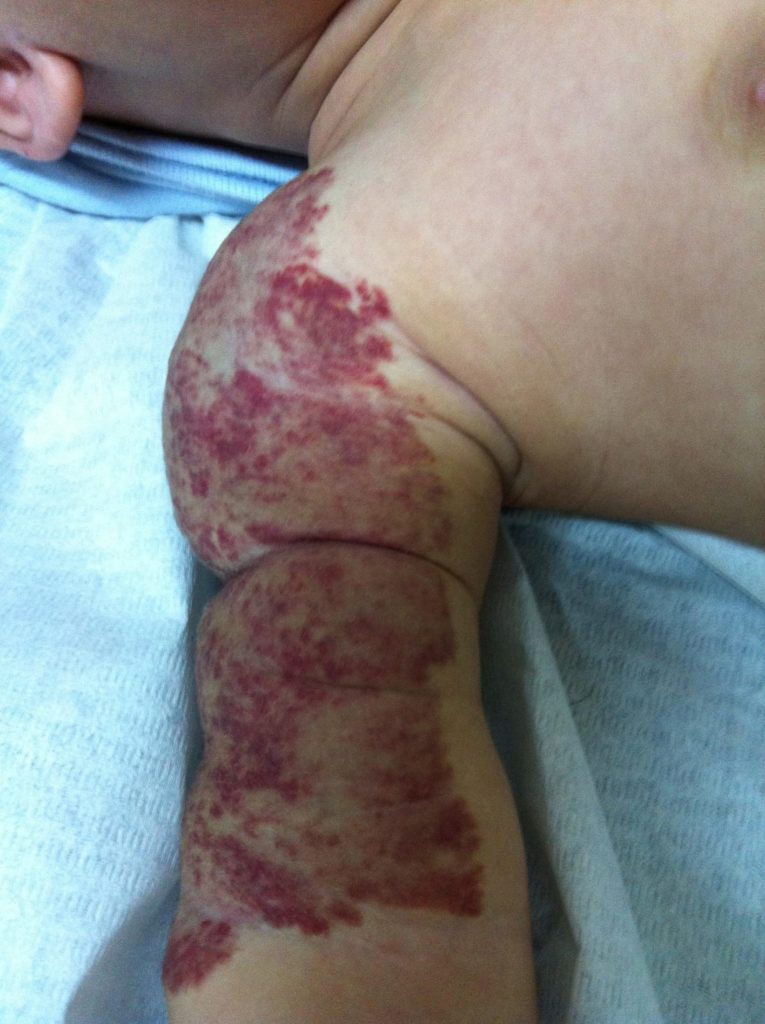
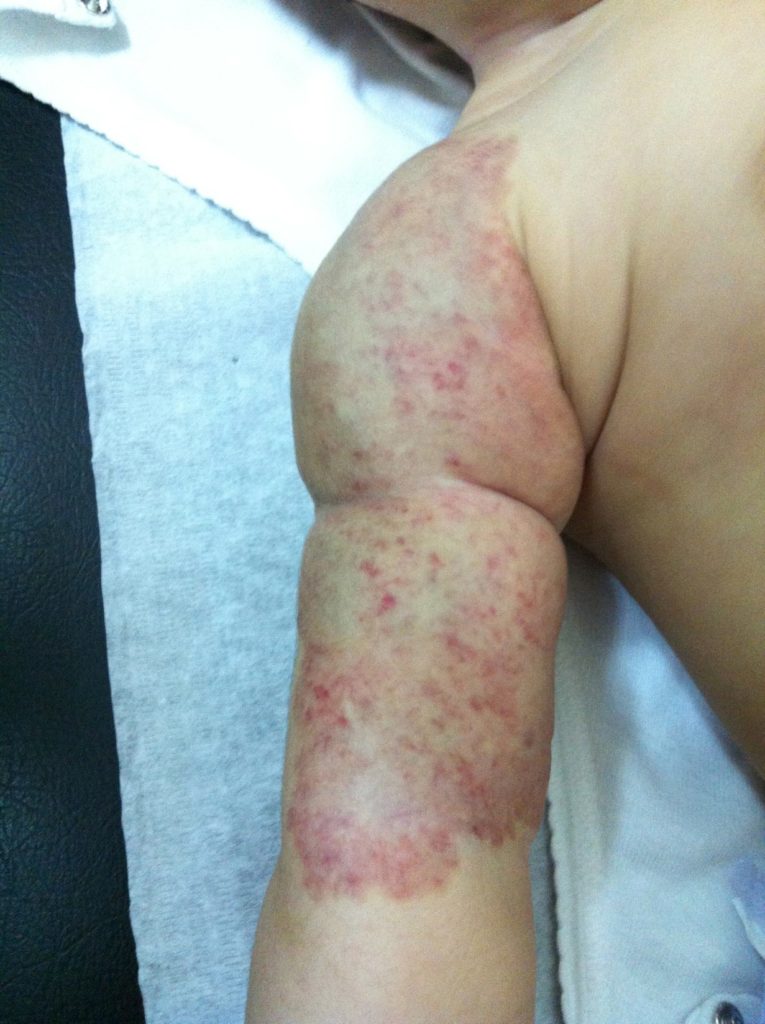
FIGURA 3. A) hemangioma segmentar antes de iniciar propranolol; B) com um mês de terapêutica; C) com três meses de terapêutica; D) com sete meses de terapêutica com propranolol
O exacto mecanismo de acção do propranolol é desconhecido, mas várias hipóteses têm sido admitidas: vasoconstrição; bloqueio de sinais proangiogénicos (vascular endothelial growth factor e basic fibroblast growth factor); regulação do sistema renina-angiotensina; inibição da produção de óxido nítrico. O propranolol parece ter um maior efeito na fase de proliferação rápida, (primeiros seis meses de vida). São contra-indicações para terapêutica com beta-bloqueante: bradicárdia sinusal, bloqueio auriculoventricular de segundo ou terceiro grau, hipotensão, choque cardiogénico, hiperreactividade brônquica e hipersensibilidade ao propranolol. Recomenda-se a realização de ECG antes de iniciar propranolol, e de ECOcardiograma quando existe suspeita de doença cardíaca. A duração da terapêutica não é consensual. A recorrência após suspensão de propranolol tem sido descrita, sobretudo antes dos 12 meses de idade. Na experiência das autoras deverá ser mantido até aos 12 meses de idade nos hemangiomas superficiais, e até aos 18 meses quando existe um componente profundo importante.
Os corticosteróides sistémicos poderão ser usados em doentes que não responderam ao propranolol ou com contra-indicação para terapêutica com beta-bloqueante. Em comparação com o propranolol, os corticóides sistémicos têm menor eficácia, são menos bem tolerados e estão associados a mais efeitos adversos.
O sirolímus (inibidor da mTOR – mammalian targert of rapamycin), com base em estudos ainda em curso e não permitindo conclusões definitivas, constitui uma nova “arma terapêutica” no âmbito dos hemangiomas complicados.
A vincristina e o interferão alfa não são recomendados como terapêutica de primeira linha devido ao seu perfil de efeitos secundários.
b) Terapêutica tópica
O timolol (um beta-bloqueante tópico) pode ser útil no tratamento dos hemangiomas infantis superficiais pequenos. Não é considerado terapêutica de primeira linha porque não estão estudados os efeitos adversos associados à absorção cutânea e porque não existe uma formulação tópica própria.
O imiquimod também parece ter algum efeito nos hemangiomas superficiais.
N.B.-De salientar que a utilização de corticóides tópicos ou intralesionais já não é recomendada.
c) Outras terapias
Inclui a crioterapia e a terapêutica laser; está praticamente reservada para os casos em que se verificam lesões cicatriciais ou telangiectasias persistentes depois da regressão do hemangioma.
Conclusão
Os hemangiomas infantis são tumores vasculares benignos, comuns no lactente e com uma história natural típica, caracterizada por uma fase de crescimento rápido, seguida de uma fase de estabilização (plateau) e de regressão espontânea até aos 4-9 anos. A terapêutica com propranolol acelera o processo de involução, diminuindo o risco de complicações, especialmente se iniciado na fase de crescimento rápido. Na experiência das autoras este tratamento deve ser reservado aos casos complicados ou em risco para tal, beneficiando de:
- diagnóstico atempado;
- referenciação precoce pelos médicos assistentes (pediatra ou médico de família);
- envolvimento duma equipa multidisciplinar incluindo, além da pediatria, dermatologia, imagiologia, oftalmologia, ORL, cardiologia pediátrica e cirurgia pediátrica.
É importante não desvalorizar o impacte psicossocial do hemangioma nas crianças e famílias, assegurá-los da sua benignidade e esclarecer a sua história natural.
BIBLIOGRAFIA
Bauland CG, Lüning TH, Smit JM, Zeebregts CJ, Sapuwen PH. Untreated hemangiomas: growth pattern and residual lesions. Plastic and Reconstructive Surgery 2011; 127: 1643-1648
Darrow Dh, Greene AK, Mancini AJ, Nopper AJ. Diagnosis and management of infantile hemangioma. Pediatrics 2015; 136: e1061-e1104
Drolet BA, Esterly NB, Frieden IJ. Hemangiomas in children. NEJM 1999; 341:173-181
Enjolras O, Gelbert F. Superficial hemangiomas: associations and management. Pediatr Dermatol 1997; 3: 173-179
Enjolras O, Mulliken JB. The current management of vascular birthmarks. Pediatr Dermatol 1993; 10: 311-313
Habif TP. Clinical Dermatology. Philadelphia: Elsevier, 2015
Haggstrom AN, Drolet BA, Baselga E, Chamlin SL, et al. Prospective study of infantile hemangiomas: demographic, prenatal and perinatal characteristics. J Pediatr 2007; 150: 291-294
Hoeger PH, Harper JI, Baselga E, Bonnet D, Boon LM, et al. Treatment of infantile haemangiomas: recommendations of a European expert group. Eur J Pediatr 2015; 174: 855-865
Kliegman RM, Stanton BF, StGeme JW, Schor NF (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2015
Krowchuk DP, Frieden IJ, Anthony J. Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics 2019; 143: e20183475; DOI: 10.1542/peds.2018-3475
Lahrichi A, Hali F, Baline K, et al. Effects of propranolol therapy in Moroccan children with infantile hemangioma. Arch Pédiatr 2018; 25: 449-451
Léauté-Labrèze C, Roque ED, Hubiche T, Boralevi F. Propranolol for severe hemangiomas of infancy. NEJM 2008; 358: 2649-2651
López-Gutiérrez JC. Clinical and economic impact of surgery for treating infantile hemangiomas in the era of propranolol: overview of single-center experience from La Paz Hospital, Madrid. Eur J Pediatr 2019; 178: 1-6
Martin JM, Sanchez S, González V, et al. Infantile hemangiomas with minimal or arrested growth: A retrospective case series. Pediatr Dermatol 2019; 36: 125-131
Melo IS, Gonçalves V, Anjos E. Propranolol nos hemangiomas infantis: casuística nacional com 30 doentes. Acta Pediatr Port 2012; 43: 139-143
Metry D, Heyer G, Hess C, et al. PHACE Syndrome Research Conference. Consensus statement on diagnostic criteria for PHACE syndrome. Pediatrics 2009; 124: 1447–1456
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Paller AS, Mancini AJ (eds). Hurwitz Clinical Pediatric Dermatology. Philadelphia: Elsevier, 2016
Starkey E, Shahidullah H. Propranolol for infantile haemangiomas: a review. Arch Dis Child 2011; 96: 890-893
Storch CH, Hoeger PH. Propranolol for infantile haemangiomas: insights into the molecular mechanisms of action. British J Dermatol 2010; 163: 269-274
Weston WL, Lane AT, Morelli JG. Color Textbook of Pediatric Dermatology. Philadelphia: Mosby Elsevier, 2007