Definição e importância do problema
A insuficiência hepática aguda (IHA) ou insuficiência hepática fulminante é uma síndroma complexa, multissistémica, que se caracteriza por alteração hepática aguda grave (necrose maciça de hepatócitos e ou alteração grave da sua função de destoxificação, síntese e excreção), subentendendo-se duração da sintomatologia inferior a 8 semanas (ver adiante).
Dum modo geral ocorre como primeira manifestação de doença em crianças previamente saudáveis, sem hepatopatia conhecida, embora também possa ocorrer naquelas com doença hepática crónica estabelecida.
Anteriormente considerava-se essencial a presença de deterioração neurológica (encefalopatia hepática) para o diagnóstico de falência hepática aguda. Actualmente, sabe-se que em idade pediátrica o compromisso neurológico, difícil de detectar em crianças pequenas, é também uma manifestação tardia no decurso da doença, não constituindo consequentemente um critério fundamental para o diagnóstico.
A prevalência da doença em idade pediátrica é desconhecida. Sabe-se que nos Estados Unidos da América (EUA) afecta até 17/100.000 pessoas/ano de todos os grupos etários e que cerca de 10 a 15% dos transplantes pediátricos são efectuados por falência hepática aguda.
Apesar de rara em pediatria, a mortalidade é elevada (até 80% em crianças não submetidas a transplante hepático). A evolução galopante da patologia implica uma minuciosa e rápida abordagem diagnóstica e terapêutica.
Etiopatogénese
Os factores etiológicos são diversos, dependendo da idade, área geográfica e nível socioeconómico. De acordo com o Quadro 1 são descritos vários grupos, salientando-se que em cerca de 40-50% dos casos não é possível identificar etiologia definida (formas ditas idiopáticas). As situações mais frequentes são a hepatite vírica, as de origem tóxico-medicamentosa e as do foro metabólico.
QUADRO 1 – Causas de insuficiência hepática aguda
| Período neonatal |
|
| Período pós-neonatal |
|
O mecanismo fisiopatológico inerente ao estabelecimento da afecção não está completamente esclarecido, nomeadamente o facto de, perante o mesmo potencial factor etiológico, somente nalgumas crianças se desenvolver o quadro clínico grave.
O quadro clínico decorre tipicamente da destruição maciça dos hepatócitos por lesão citotóxica directa ou por resposta imunogénica. Existem dois tipos de lesão principal: 1) – necrose hepática extensa com colapso da arquitectura lobular (mais comum na doença provocada por vírus hepatotrópicos, intoxicação por paracetamol ou intoxicação por cogumelos); 2) – degenerescência hepatocelular com esteatose maciça e necrose hepática pouco extensa (situação mais frequentemente associada a doenças metabólicas).
Outros factores associados à lesão do hepatócito incluem: alteração do processo de regeneração, hipoperfusão sanguínea do parênquima, endotoxémia, e depressão da função do SRE.
É frequente surgir coagulopatia, a qual ocorre por diminuição da síntese hepática de factores de coagulação, associada a um aumento do consumo dos mesmos assim como de plaquetas.
A hipoglicemia, presente em 40% dos doentes, surge associada sobretudo a diminuição da neoglicogénese e das reservas de glicogénio, paralelamente a um aumento das necessidades em glucose, e a hiperinsulinémia por diminuição da depuração hepática.
O mecanismo da encefalopatia pode relacionar-se com a hiperamoniémia, incremento da actividade dos receptores de GABA e incremento de níveis circulantes de compostos endógenos formados, semelhantes a benzodiazepinas; todos estes produtos têm o seu processo de depuração hepática comprometida, num círculo vicioso.
Reportando-nos ao Quadro 1, são abordados os principais factores etiológicos:
Infecções
As infecções são a causa mais frequente de insuficiência hepática aguda, variando o agente etiológico consoante o grupo etário e a área geográfica. Em regra, aponta-se a proporção de 1-2% de hepatites víricas em geral evoluindo para IHA.
É consensual que agentes como os vírus da hepatite A, da hepatite B, vírus herpes simplex, parvovírus B16, enterovírus e adenovírus podem provocar IHA.
Existem outros microrganismos que poderão originar lesão hepática mas, isoladamente, raramente provocam falência hepática aguda, nomeadamente: vírus da hepatite C/VHC, vírus de Epstein Barr/VEB, citomegalovírus/CMV, herpes vírus 6 e vírus da imunodeficiência humana/VIH.
Nos países ditos desenvolvidos, 80% dos casos de IHA são provocados por hepatite, sintomática em menos de 1% das crianças; esta baixa proporção tem tendência a diminuir tendo em conta a imunização levada a cabo nalguns países.
A hepatite B é uma causa importante de falência hepática em países em desenvolvimento nos países em que a doença é endémica e não existe programa de imunização apropriado. A mortalidade é maior em adultos jovens e quando a transmissão ocorre fora do período perinatal. Relativamente à comparticipação da infecção por vírus da hepatite E, rara nos países desenvolvidos, é uma das principais causas de IHA no subcontinente indiano.
A anamnese e o exame objectivo são essenciais, já que poderão fazer suspeitar do agente etiológico causal; da apresentação clínica poderão fazer parte manifestações clínicas e/ou laboratoriais apontando para determinado agente: é o caso da varicela, da infecção por vírus de Epstein Barr ou por Parvovírus B19 (cursando este último frequentemente com anemia aplásica).
Fármacos e tóxicos
Numerosos fármacos e substâncias constituem a segunda causa mais frequente de IHA nas crianças, tendencialmente com melhor prognóstico do que a doença provocada por infecções. A lesão hepática ocorre por mecanismo hepatotóxico directo ou por reacção idiossincrática, conforme a lesão seja dependente da dose ou não.
Pela sua utilização muito frequente em pediatria, o paracetamol é uma das causas mais frequentes de IHA. Esta pode ocorrer associada: – à ingestão aguda de doses superiores a 100mg/kg/dia (tipicamente, ingestão intencional nos adolescentes ou, mais raramente, ingestão acidental em crianças pequenas): – ou à ingestão crónica de doses superiores a 15mg/kg/dose a cada 4 horas durante mais de 24 a 48 horas. Têm maior risco de hepatotoxicidade as crianças mais novas, com período de jejum prolongado ou as que são medicadas concomitantemente com outros fármacos potencialmente hepatotóxicos (anticonvulsantes, isoniazida, entre outros).
A seguir ao paracetamol, os fármacos implicados com mais frequência são a isoniazida, o propiltiouracilo, a fenitoína e o ácido valpróico.
Para além dos fármacos, importa salientar a toxicidade conhecida de longa data provocada pela ingestão de toxinas através do cogumelo Amanita phalloides.
Hepatopatias autoimunes e doenças neoplásicas infiltrativas
As hepatopatias autoimunes são causas raras de IHA, acompanhando-se de autoanticorpos positivos (ANA e antimúsculo liso) e hipergamaglobulinémia.
Quanto às doenças hemato-oncológicas salientam-se a linfo-histiocitose hemofagocítica, as leucemias e os linfomas; trata-se de afecções com prognóstico reservado ou mau, em cuja base etiopatogénica está uma infiltração maciça do fígado.
Doenças metabólicas
De acordo com a literatura, as causas metabólicas comparticipam na proporção de 10% dos casos de insuficiência hepática aguda nos EUA e Europa do Norte.
Geralmente a falência hepática de causa metabólica ocorre em recém-nascidos ou lactentes pequenos em contexto de doença sistémica, muitas vezes sem diagnóstico etiológico prévio. Torna-se óbvio que será da maior importância o diagnóstico etiológico feito em fase precoce, tendo em conta que a doença de base poderá ter tratamento específico, o que poderá conduzir a regressão da hepatopatia.
As afecções do foro metabólico, designadamente as que evoluem para IHA, para além do grave compromisso do estado geral, cursam geralmente com hipoglicemia, acidose e aumento do ácido láctico. Ao contrário da maioria das outras causas de insuficiência hepática, tipicamente existe hepatomegalia.
No período neonatal e no pequeno lactente com insuficiência hepática aguda devem ser ponderadas etiologias como a galactosemia, a tirosinémia, a doença de Niemann-Pick tipo C e ainda doenças mitocondriais. Nas crianças mais velhas e adolescentes a IHA pode estar associada a erros inatos da síntese de ácidos biliares, intolerância hereditária à frutose, doença de Wilson ou doenças da oxidação dos ácidos gordos, entre outras causas.
Outras causas
Por fim, a lesão hepática grave poderá constituir um epifenómeno de hipoperfusão hepática por problema circulatório, como, por exemplo, choque grave como acontece em doentes com quadro de sépsis ou pós-hemorragia aguda.
Também, toda e qualquer situação em que haja obstrução na saída do fluxo venoso hepático (designadamente na doença vasoclusiva e síndroma de Budd-Chiari) poderá conduzir a IHA.
Manifestações clínicas
Como regra geral, pode referir-se que a apresentação clínica depende do factor causal.
Por outro lado, a falência hepática (aguda/fulminante) em idade pediátrica poderá ser:
- a primeira manifestação de doença hepática; ou
- a complicação de doença hepática previamente conhecida (situação designada “aguda sobre crónica”).
Na segunda condição (2.) poderá verificar-se previamente atraso do neurodesenvolvimento ou disfunção neuromuscular, quadro provavelmente relacionado com doença mitocondrial, ou doença da beta-oxidação.
Os sinais e sintomas associados a IHA surgindo de modo progressivo, de duração inferior a 8 semanas, são: icterícia, fetor hepático, anorexia, febre, náuseas, vómitos, hipoglicemia, dor abdominal, culminando em diátese hemorrágica (epistaxes, gengivorragias, equimoses fáceis, hemorragia abundante nos locais de punção venosa ou, nos casos mais graves, hemorragia digestiva), ascite e, depois, diminuição das dimensões do fígado (sendo este último sinal de muito mau prognóstico).
Esta evolução progressiva poderá ser acompanhada de dificuldade alimentar, e doutra sintomatologia relacionada com quadro de encefalopatia cuja gravidade depende do grau de edema cerebral. É a principal causa de mortalidade.
Sistematizando a gravidade da encefalopatia através de determinados sinais e sintomas, e de alterações do EEG, são descritos 4 graus: I- orientação espacial alterada e alterações mínimas do EEG; II- confusão, letargia ou agitação, associada a EEG lento generalizado; III- Estupor e hiperreflexia, com EEG lento anormal; IV- coma associado a hipertonia inicialmente, seguindo-se fase de hipotonia, arreflexia osteotendinosa, óculo-cefálica e pupilar, com EEG evidenciando ondas delta. Ulteriormente verifica-se postura típica dos estados de descerebração e de descorticação.
A encefalopatia hepática, mais prevalente em crianças abaixo dos três anos de idade, ocorre em fase mais tardia da evolução atrás descrita; pode traduzir-se por convulsões, por alterações subtis, inespecíficas, do comportamento, ou ainda por períodos de desorientação discreta.
Em crianças mais velhas, em certas formas clínicas de IHA idiopática, a encefalopatia poderá manifestar-se cerca de 8-24 semanas após o início da icterícia.
Em suma, tendo em conta a necessidade de elevado nível de suspeição por parte do clínico, haverá que valorizar determinados sinais e sintomas sugestivos de doença hepática que podem sugerir o diagnóstico de IHA, nomeadamente, coagulopatia, hipoglicémia e encefalopatia.
Diagnóstico
Não existem critérios diagnósticos estabelecidos para a IHA na idade pediátrica.
A encefalopatia, habitualmente presente, não constitui, no entanto, um critério obrigatório porque o seu diagnóstico em idade pediátrica é difícil, sobretudo nas crianças mais pequenas; por outro lado, tal problema é mais notório na fase terminal da doença.
Tipicamente, a suspeita de falência hepática recai na criança sem patologia prévia, com um início agudo (menos de 8 semanas) de icterícia, anorexia e mal-estar geral, alterações bioquímicas da função hepática, coagulopatia (com tempo de protrombina superior a 15 segundos ou INR > 1,5 não corrigido com a administração de vitamina K, na presença de encefalopatia; ou > 20 segundos com INR > 2 na situação com ou sem encefalopatia) e com alterações do estado de consciência.
É essencial uma história clínica minuciosa, designadamente, no que diz respeito a: data de início e duração dos sintomas, exposição a contactos com hepatite infecciosa, história de transfusões sanguíneas prévias e de medicação (nomeadamente sem prescrição médica), e a história familiar de hepatite infecciosa, autoimune ou outras doenças sistémicas com envolvimento hepático.
Nos recém-nascidos é importante a revisão de possíveis infecções do grupo TORCH e, nos adolescentes, a exposição a drogas, tatuagens ou piercings.
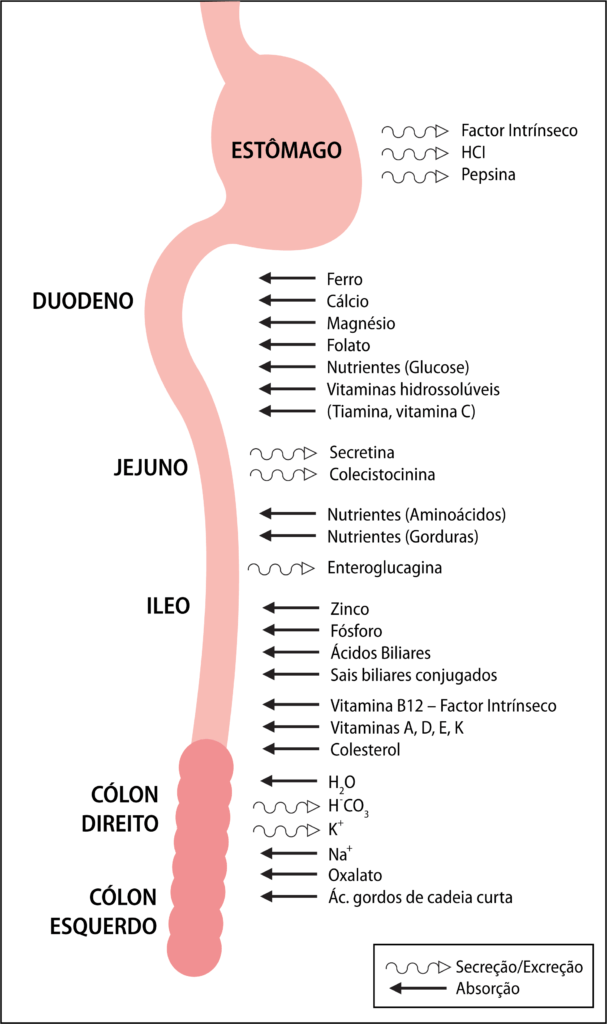
No Quadro 2 são descritos os exames complementares a realizar numa primeira abordagem da criança com IHA. Deve ser analisada a enzimologia hepática a fim de aferir o grau de infecção, inflamação e lesão hepáticas: como regra, existe elevação da alanina-aminotransferase e aspartato-aminotransferase assim como da bilirrubina sérica. A síntese hepática de albumina, colesterol e ureia encontra-se diminuída, existindo geralmente hiperamoniémia por diminuição da depuração da amónia.
QUADRO 2 – Avaliação da insuficiência hepática aguda
*Diagnóstico de coagulopatia; 1) na presença de encefalopatia hepática: INR > 1,5 e TP = ou > 15 segundos; 2) com ou sem encefalopatia hepática: INR > 2 e TP = ou > 20 segundos |
| Investigação geral |
Exames Laboratoriais (sangue)
Exames imagiólogicos
Exame neurofisiológico
|
| Investigação etiológica |
|
As alterações da coagulação ocorrem em 100% das crianças com IHA, salientando-se que valor de factor V inferior a 17%, de factor VII inferior a 8%, e INR superior a 4 são sinais de mau prognóstico.
Sempre que possível, deve tentar-se identificar a causa da insuficiência hepática, tendo em vista a eventualidade de se dispor de terapêutica específica em função da respectiva etiologia.
A investigação etiológica e os exames complementares a realizar deverão ter em conta o grupo etário e a suspeita diagnóstica, entre outros factores. São exemplos as investigações laboratoriais seguintes:
- serologias para vírus (VHA, VHB, RNA VHC, CMV, VIH, parvovírus B19 ou adenovírus);
- pesquisa de tóxicos (nomeadamente, pela frequência, o paracetamol); e
- pesquisa de marcadores de autoimunidade como os anticorpos antinucleares/ANA e antimúsculo liso/AML ou SMA.
A biópsia hepática, não essencial, não é normalmente efectuada nas crianças com IHA tendo em consideração o risco de hemorragia, e o valor limitado da histologia nas estratégias diagnóstica e terapêutica. Habitualmente, a biópsia fica reservada para: – doentes com suspeita de doença de Wilson, caso os outros critérios diagnósticos sejam inconclusivos, ou; – situações associadas a etiologia indeterminada.
Tratamento
O tratamento da falência hepática aguda compreende: – medidas gerais; – medidas específicas consoante a etiologia; e – outras medidas extraordinárias como o transplante hepático urgente.
Dada a gravidade e a morbimortalidade elevadas, todas as crianças devem ser admitidas em unidades de cuidados intensivos pediátricos com acesso a programa de transplante hepático.
A terapêutica traduz-se essencialmente pelas seguintes medidas: – evitar as complicações; – tratar a encefalopatia hepática; – evitar factores potencialmente desencadeantes ou agravantes (sobrecarga hídrica, sobrecrescimento bacteriano, profilaxia da hemorragia gastrintestinal); e – controlar a coagulopatia (designadamente através da administração de vitamina K e de factores de coagulação).
Importa salientar que na intoxicação por paracetamol deve ser utilizada a N-acetilcisteína como antídoto e, quando a causa da insuficiência hepática é a infecção por herpes vírus ou citomegalovírus, deve ser efectuada terapêutica antivírica específica (aciclovir e ganciclovir, respectivamente).
Existem actualmente sistemas de suporte hepático artificial com depuração extra-hepática dos metabólitos, susceptíveis de substituição temporária da função hepática.
A transplantação hepática é, no entanto, a única medida curativa disponível actualmente nos doentes com IHA. Deverá ser realizada o mais precocemente possível, salientando-se que o dano neurológico irreversível com edema ou herniação cerebral, a falência multiorgânica, e a sépsis são contraindicações formais para a sua realização.
Prognóstico
O prognóstico das crianças com IHA é reservado, com uma mortalidade que atingia, na era pré-transplante, até 80% das crianças. Actualmente, a sobrevivência aumentou, o que se deve aos progressos realizados no campo dos cuidados intensivos pediátricos, e à rápida inserção destas crianças em centros de referência com vasta experiência e programas de transplante hepático.
A sobrevivência espontânea, sem recurso ao transplante hepático, é maior nas crianças com intoxicação por paracetamol (até 94%), e menor nas com doença metabólica associada.
São factores de mau prognóstico: – a etiologia (nomeadamente se insuficiência associada a fármacos ou a hepatite não A); – a idade (crianças com idade inferior a 2 anos); – a presença de encefalopatia grave; – a apresentação clínica subaguda ou fulminante; e – bilirrubina sérica superior a 17,5mg/dL.
É importante referir que, mesmo nas crianças submetidas a transplante hepático por falência hepática aguda, a sobrevivência global é de apenas de 74% no final do primeiro ano, e de 69% após quatro anos. A sobrevivência é maior nas crianças submetidas a transplante por doença hepática crónica, sem falência aguda.
BIBLIOGRAFIA
Cochran JB, Losek JD. Acute liver failure in children. Pediatric Emergency Care 2007; 23:129-135
Coelho T, Tredger M, Dhawan A. Current status of immunosuppressive agents for solid organ transplantation in children. Pediatr Transplant 2012; 16:106-122
Craig DGN, Lee A, Hayes PC, et al. Review article: the current management of acute liver failure. Aliment Pharmacol Ther 2010; 31: 345 – 358
Devictor D, Tissieres P, Durand P, et al. Acute liver failure in neonates, infants and children. Expert Rev Gastroenterol Hepatol 2011; 5: 717 – 729
Dhawan A, Cheeseman P, Mieli-Vergan IG. Approaches to acute liver failure, in children. Pediatr Transplant 2004; 8: 584-588
Gómez JMJ, Miquel BP, Aliaga, ED. Fallo hepático agudo. Protocolos diagnóstico-terapéuticos de Gastroenterología, Hepatología y Nutrición Pediátrica SEGHNP-AEP, 2010
Kelly DA (ed). Diseases of Liver and Biliary System in Children. London: Blackwell Science, 2008
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education,
Mohammad S, Alonso EM. Approach to optimizing growth, rehabilitation, and neurodevelopmental outcomes in children after solid-organ transplantation. Pediatr Clin North Am. 2010; 57:539-557
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Shawcross D, Jallan R. Dispelling myths in the treatment of hepatic encaphalopathy. Lancet 2005; 265:431- 433
Squires RH, Dhawan A, Alonso E, et al. Intravenous Nacetylcysteine in pediatric patients with nonacetaminophen acute liver failure. Hepatology 2013; 57: 1542-1549
Sundaram V, Shneider BL, Dhawan A, et al. King’s College Hospital criteria for non-acetaminophen induced acute liver failure. J Pediatr 2013; 162:319-323
Venick RS, Farmer DG, McDiarmid SV, et al. Predictors of survival following liver transplantation in infants: a single-center analysis of more than 200 cases. Transplantation 2010; 15, 89:600-605
Walker WA, Goulet O, Kleinman RE (eds). Pediatric Gastrointestinal Disease. Hamilton, Ontario: Decker, 2004
Willie R, Hyams JS, Kay M (eds). Pediatric Gastrointestinal and Liver Disease. Philadelphia: Elsevier, 2016