Sistematização e importância do problema
Um grupo importante de displasias esqueléticas integra entidades clínicas devidas a alterações dos receptores transmembrana, entre as quais se encontram as condrodisplasias associadas a defeitos do receptor 3 do factor de crescimento de fibroblastos (FGFR3) e a defeitos do receptor da PTH (ver atrás).
Entre as displasias relacionadas com mutações em diferentes loci no gene FGFR3 (sendo que a localização da mutação se correlaciona com o fenótipo) incluem-se:
- A displasia tanatófora de tipo I e II – a condrodisplasia letal mais comum, com uma prevalência de 1/35.000 nascimentos;
- A acondroplasia, a displasia esquelética/condrodisplasia, não letal, mais frequente, evidenciando prevalência de 1/15.000 a 1/40.000 nascimentos; e
- A hipocondroplasia.
Neste capítulo procede-se a uma abordagem sucinta das referidas condrodisplasias (1., 2., e 3.) dando ênfase à acondroplasia e a aspectos do diagnóstico diferencial.
1. DISPLASIA TANATÓFORA
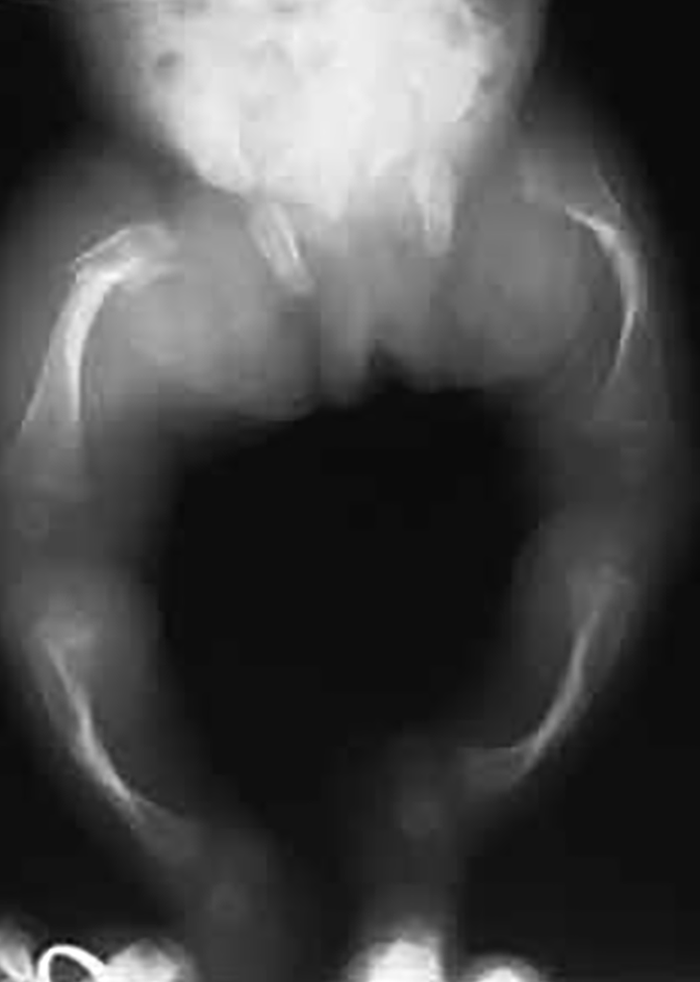
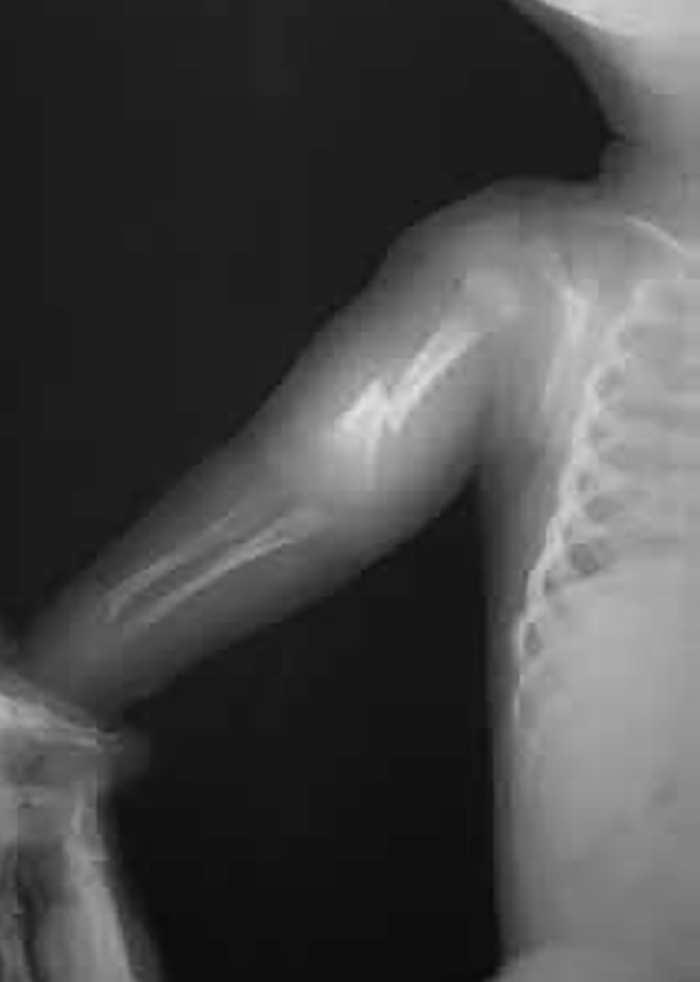
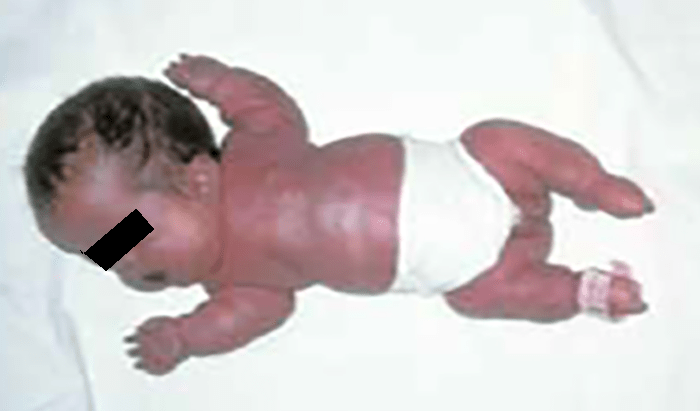
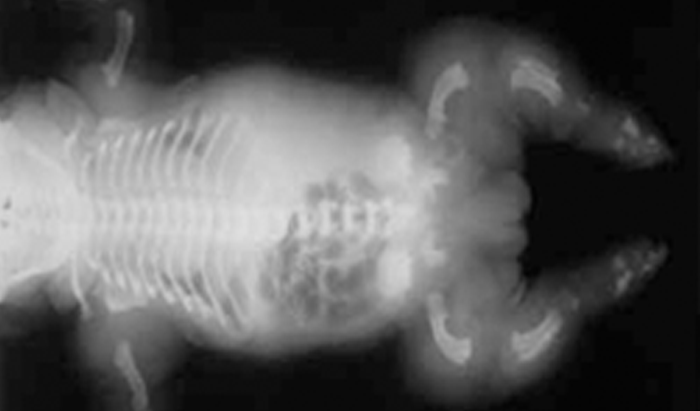
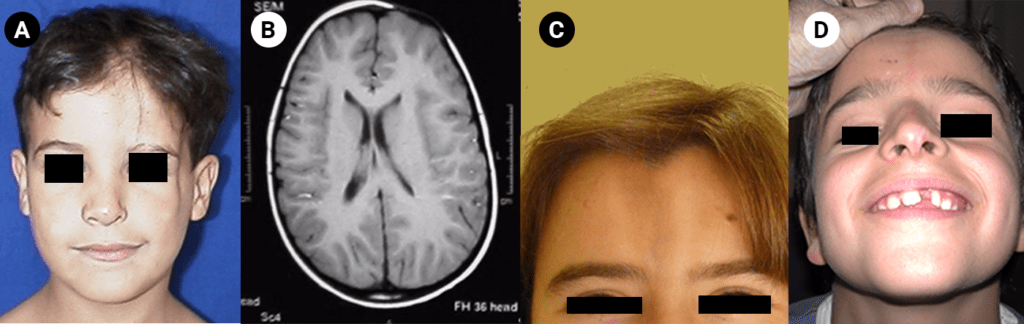
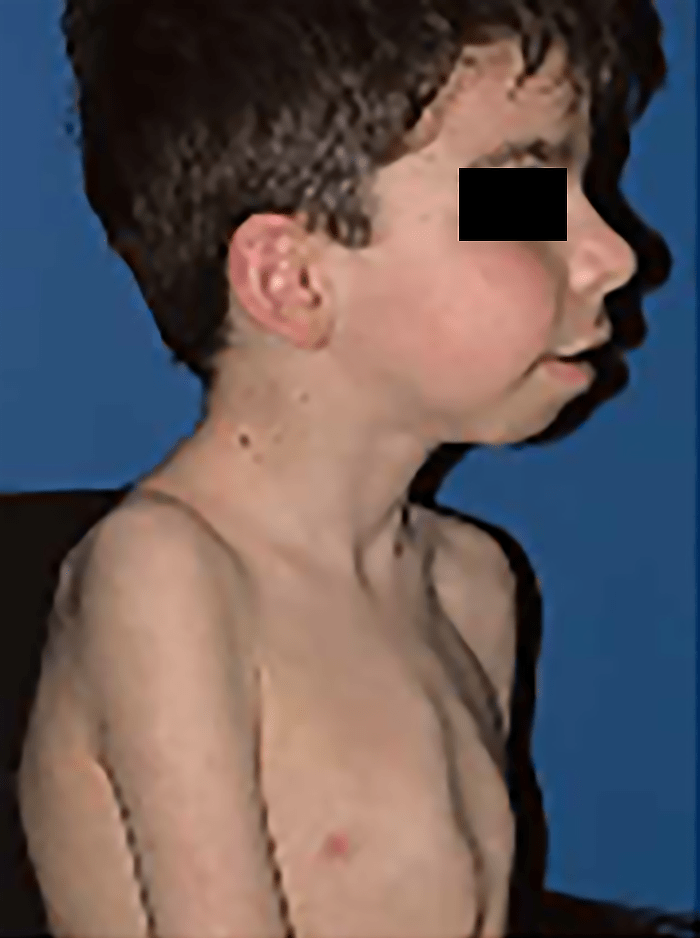
Trata-se duma displasia grave, em geral letal no pré-parto (geralmente pré-termo) ou nas primeiras horas de vida, tendo decorrido a gravidez com poli-hidrâmnio. No exame físico ressalta fenótipo peculiar caracterizado essencialmente por macrocefalia, por vezes deformação craniana em “folha de trevo”, hipoplasia média facial, pescoço curto, tórax longo e estreito “em sino”, e membros muito curtos.
A destrinça entre tipo I e tipo II decorre de padrões radiográficos esqueléticos específicos. Sob os pontos de vista genético e biomolecular, cabe referir que no tipo I a mutação se situa no códon 650, com substituição de lisina por ácido glutâmico, enquanto no tipo II se associa a deleção da porção terminal carboxílica do FGFR3.
Diagnóstico diferencial
O diagnóstico diferencial principal faz-se com a acondrogénese, especialmente na forma homozigótica.
Nesta última situação a cabeça é de maiores dimensões, a ossificação é mais deficiente nos corpos vertebrais, especialmente na região lombar, verificando-se ausência no sacro, púbis, ílio e ísquio.
2. ACONDROPLASIA
Etiopatogénese e genética
A acondroplasia é o protótipo das condrodisplasias.
Todos os doentes com a acondroplasia típica têm mutações no gene FGFR3, códon 380 (arginina substituída por glicina), sendo a transmissão do tipo autossómico dominante; na sua maioria, os casos decorrem de uma nova mutação de progenitores normais.
Uma vez que, entre a população adulta com nanismo a acondroplasia é relativamente frequente, e frequentes são os casamentos entre tais pacientes, existe o risco hereditário de transmitir a cada descendente a doença (risco de 50% para as formas heterozigóticas, e 25% para as formas homozigóticas, estas últimas geralmente letais no período neonatal).
Manifestações clínicas
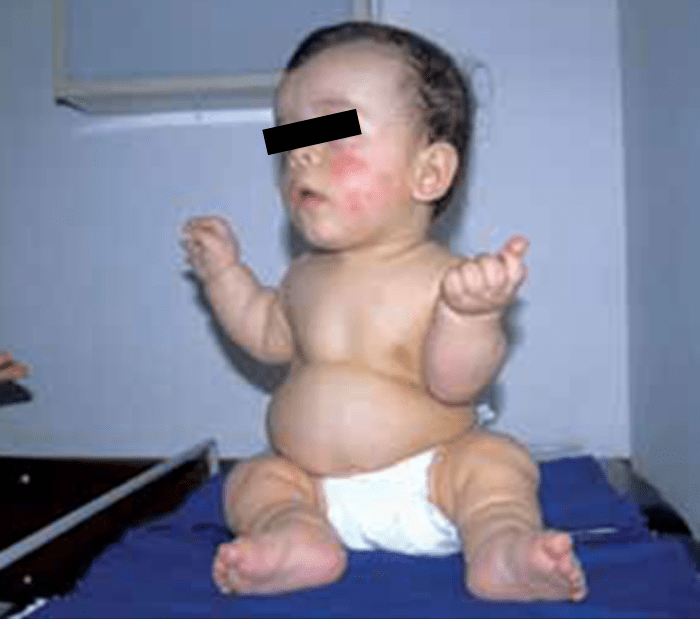
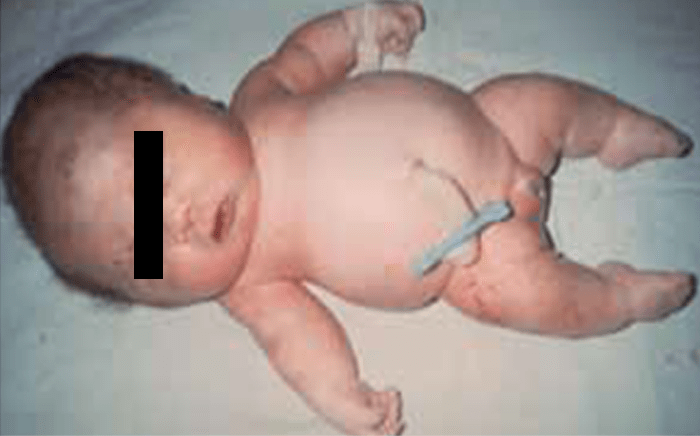
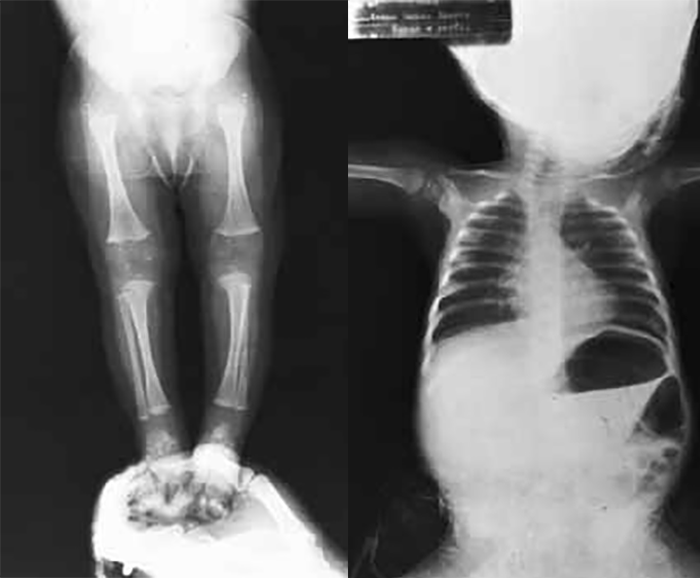
Tipicamente, o recém-nascido tem membros curtos, tronco alongado e estreito, macrocefalia com fronte proeminente, fontanela grande e nariz curto e dentes apinhados.
As alterações vertebrais manifestam-se por estreitamento do canal medular ao nível da coluna cervical e lombar. A gibosidade lombar que se verifica nas primeiras idades é substituída, com o avançar dos anos, por lordose lombar.
As alterações cranianas podem acompanhar-se de hidrocefalia em 6% dos casos, e de compressão ao nível do buraco occipital (5-10% dos casos) com risco de apneia central e morte súbita. Por vezes tal patologia pode associar-se a instabilidade da coluna cervical.
São comuns os quadros de hipoacusia e otite média recorrente associados a estenose do cavum, o que por vezes poderá estabelecer indicação para amigdalectomia.
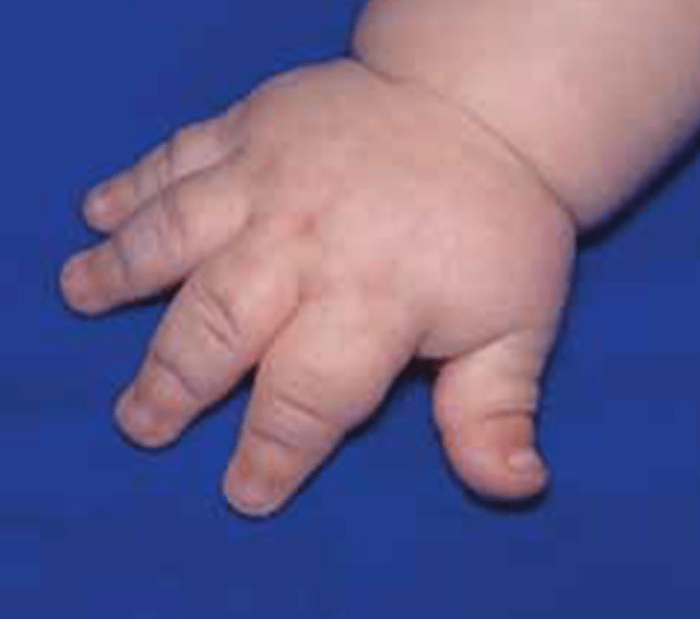
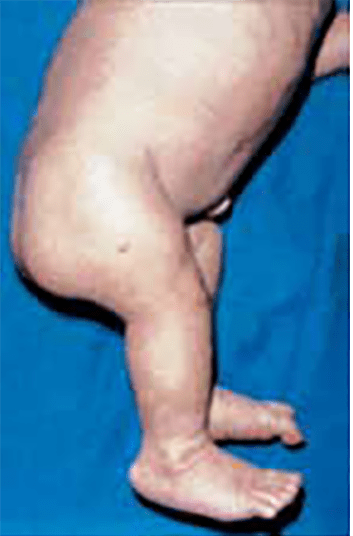
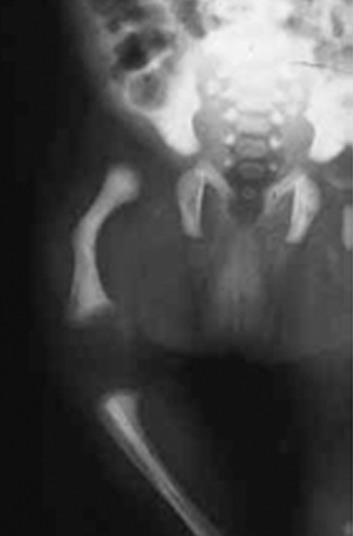
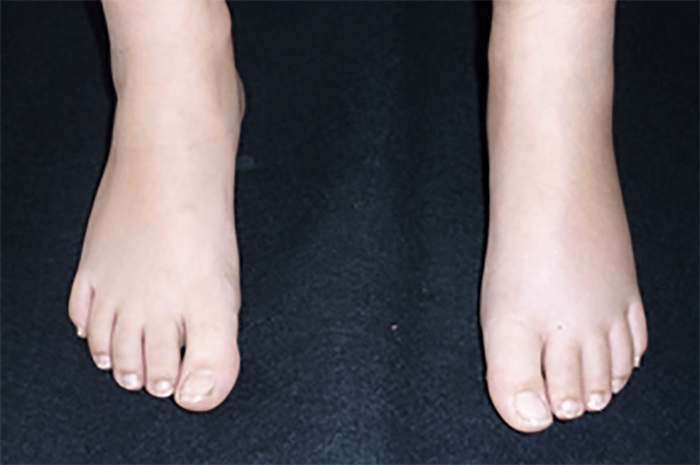
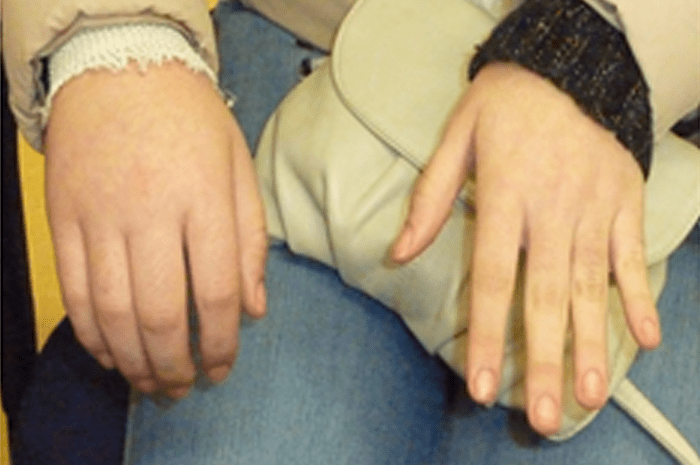
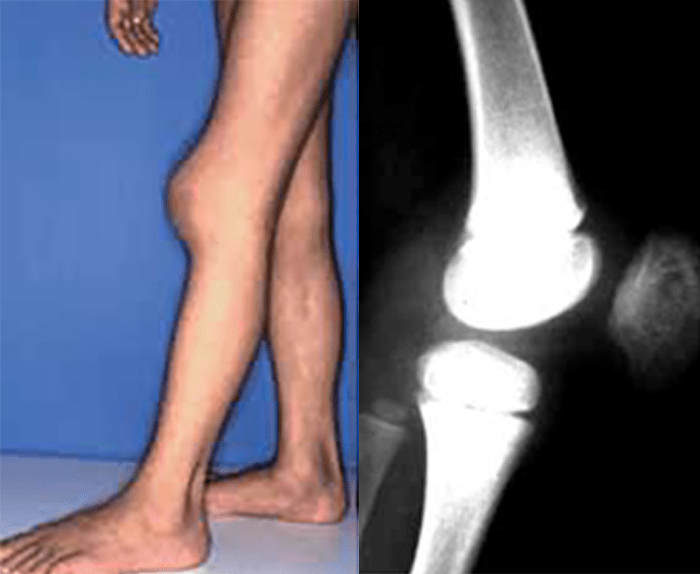
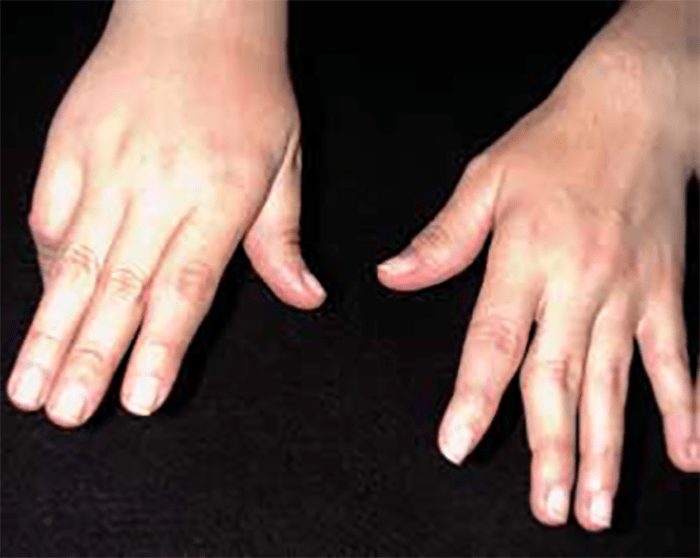
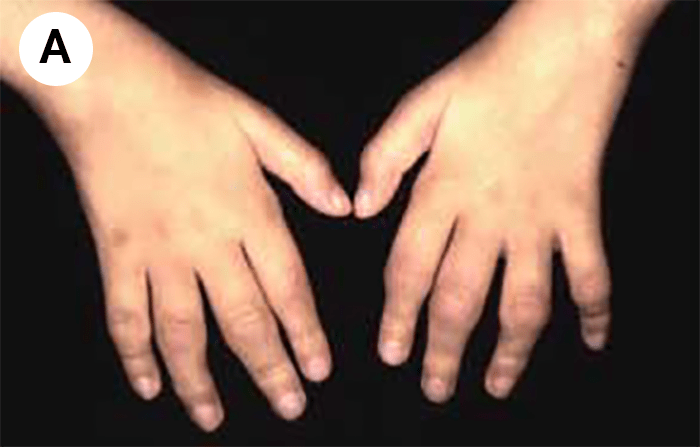
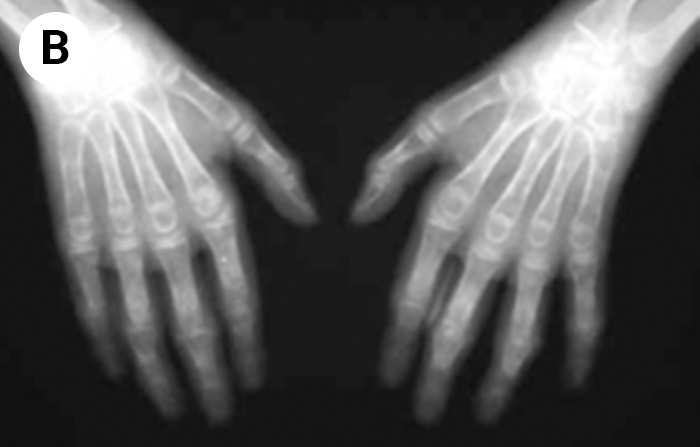
O encurtamento dos membros é mais acentuado nos segmentos proximais e os dedos exibem muitas vezes uma configuração em tridente. O encurvamento mais acentuado da tíbia é notório, ao mesmo tempo que se verifica que o comprimento do perónio é desproporcionalmente mais comprido do que a tíbia.
Verifica-se hiperextensibilidade articular, embora ao nível do cotovelo se verifique restrição na extensão. Geralmente o comprimento ao nascer é anormalmente reduzido ou, por vezes, está no limite inferior do normal. Em regra, na idade adulta, a estatura é cerca de 118-145 cm no sexo masculino, e 112-136 cm no sexo feminino.
A inteligência é normal, excepto nos casos complicados de lesões neurológicas graves.
Diagnóstico diferencial
O diagnóstico diferencial faz-se essencialmente com outras formas de displasia com membros curtos, tais como, as displasias tanatófora, acondrogénese, metatrópica, torácica asfixiante, diastrófica, hipocondroplasia e síndroma de Ellis-van Creveld.
Actuação prática
A prestação de cuidados a pacientes com a patologia em análise implica um apoio multidisciplinar. De acordo com determinada sintomatologia específica, e com a coordenação por pediatra ou médico de família, poderão estar indicadas certas intervenções invasivas no âmbito da ORL, ortopedia e neurocirurgia.
Em centros especializados, com o apoio do endocrinologista, têm sido utilizados (de forma não consensual entre especialistas) fármacos tais como a GH (hormona de crescimento recombinante) e um análogo do péptido natriurético do tipo C como medidas para estimular o crescimento ósseo.
3. HIPOCONDROPLASIA
A hipocondroplasia partilha características clínicas e radiológicas esqueléticas com a acondroplasia, mas em menor grau e é mais ligeira. As dismorfias faciais são mais suaves, sendo que por vezes apenas é notória a proeminência frontal. Na idade adulta, em média são atingidas estaturas da ordem de 140 cm no sexo masculino e de 135 cm no sexo feminino.
Caracterizando-se esta condrodisplasia por muita heterogeneidade genética, cabe referir que em cerca de 3/4 dos pacientes foram detectadas alterações no gene FGFR3, correspondendo a mutação mais comum a alteração no códon 470; contudo, segundo a literatura, foram também detectadas anomalias ao nível do códon 540.
Diagnóstico diferencial
O diagnóstico diferencial faz-se essencialmente com a acondroplasia, pela sua expressão clínica e radiológica menos marcada. Com efeito, o fenótipo facial é aparentemente normal na hipocondroplasia, o que não acontece naquela.
Actuação prática
Quanto a actuação terapêutica do foro médico, nalguns centros tem sido utilizada a GH recombinante, a exemplo do que foi referido a propósito da acondroplasia.
BIBLIOGRAFIA
Carter EM, Raggio CL. Genetic and orthopedic aspects of collagen disorders. Curr Opin Pediatr 2009; 21: 46-54
Chitty LS, Tan AW, Nesbit DL, et al. Sonographic diagnosis of SEDC and double heterozygote of SEDC and achondroplasia – a report of six pregnanacies. Prenat Diagn 2006; 26: 861-865
Goodman RM, Gorlin RJ. The Malformed Infant and Child. An Illustrated Guide. Oxford: Oxford University Press, 1983
Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet 2007; 370: 162-172
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Krakow D. Skeletal dysplasias. Clin Perinatol 2015; 42: 301-319
Krakow D, Alanay Y, Rimoin LP, et al. Evaluation of prenatal onset osteochondrodysplasias by ultrasonography: a retrospective and prospective analysis. Am J Med Genet A 2008; 146A: 1917-1924
Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med 2009; 11: 127-133
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Parilla BV, Leeth EA, Kambich MP, et al. Antenatal detection of skeletal dysplasias. J Ultrasound Med 2003; 22: 255-258
Pombo M, et al (eds). Tratado de Endocrinologia Pediátrica. Madrid: McGraw-Hill Interamericana, 2010
Ross JL, Bellus G, Scott CI, et al. Mesomelic and rhizomelic short stature: the phenotype of combined Leri-Weill dyschondrosteosis and achondroplasia or hypochondroplasia. Am J Med Genet A 2003; 116: 61-65
Salmon MA, Lindebaum RH. Developmental Defects and Syndromes. Aylesbury: HM+M Publishers, 1978
Savarirayan R, et al. C-type natriuretic peptide analogue therapy in children with achondroplasia. NEJM 2019; 381: 25-38
Smith DW. Recognizable Patterns of Human Malformation. Philadelphia: Saunders, 1982
Trotter TL, Hall JG. Health supervision for children with achondroplasia. Pediatrics 2005; 116: 771-783
Unger S. A genetic approach to the diagnosis of skeletal dysplasia. Clin Orthop Relat Res 2002; 401: 32-38
Warman ML, Cormier-DaireV, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 2011; 155A: 943-968
Wright MJ, Irving MD. Clinical management of achondroplasia. Arch Dis Child 2012; 9: 129-134