Definição e importância do problema
As vasculites constituem um grupo heterogéneo de doenças, na sua globalidade raras, de etiopatogénese não totalmente esclarecida, caracterizadas por inflamação e necrose da parede dos vasos, do que resulta grau variável de isquémia e lesão teciduais ao nível de diversos territórios.
Todo e qualquer tipo de vaso e toda e qualquer estrutura do mesmo vaso podem ser afectados; salienta-se que diferentes estruturas podem ser afectadas de modo selectivo, o que condiciona manifestações clínicas muito variáveis.
Assim, tal patologia pode classificar-se atendendo a critérios diversos, compreendendo-se que determinada entidade possa estar incluída em mais do que uma categoria classificativa:
- Topográfica
Localizadas (restritas à pele) ou sistémicas (envolvendo órgãos internos); - Etiológica
Primárias ou secundárias (associadas principalmente a infecções, a doenças reumáticas como artrite idiopática juvenil, lúpus eritematoso sistémico, dermatomiosite juvenil e a neoplasias); - Histológica
Leucocitoclástica (púrpura de Henoch-Schönlein, vasculite de hipersensibilidade), necrosante (poliarterite nodosa – PAN, doença de Kawasaki), granulomatosa (granulomatose com poliangeíte e granulomatose eosinofílica com poliangeíte), de células gigantes (arterite de Takayasu) e outras (doença de Behçet); - Anátomo-fisiológica (calibre dos vasos)
Afectando vasos grandes/aorta e ramos proximais (arterite de Takayasu), vasos médios/artérias renais, coronárias e vasculatura mesentérica (doença de Kawasaki, poliarterite nodosa) e vasos pequenos/capilares, arteríolas e vénulas pós-capilares (púrpura de Henoch-Schönlein, vasculite de hipersensibilidade, poliangeíte microscópica, granulomatose com poliangeíte e granulomatose eosinofílica com poliangeíte); - Associadas ao anticorpo dirigido contra antigénio do citoplasma de neutrófilos (ANCA)
Este grupo baseia-se na presença ou ausência de anticorpo dirigido contra antigénio do citoplasma de neutrófilos (ANCA, sendo pANCA-perinuclear, e cANCA- citoplásmico). As vasculites ANCA positivo são: granulomatose com poliangeíte, granulomatose eosinofílica com poliangeíte e poliangeíte microscópica.
O diagnóstico de vasculite baseia-se:
- Na anamnese e no exame objectivo pormenorizados (designadamente investigando eventual utilização de fármacos, palpação de pulsos, e medição da pressão arterial);
- Em exames complementares seleccionados em função da história clínica, os quais poderão culminar no exame histopatológico (biópsia) e no estudo de biologia molecular.
Existem vários achados clínicos sugestivos de vasculite (Quadro 1).
QUADRO 1 – Achados clínicos sugestivos de vasculite
|
O Quadro 2 integra alguns parâmetros laboratoriais que, embora inespecíficos, sugerem a presença de vasculite.
QUADRO 2 – Parâmetros laboratoriais possivelmente associados a vasculite
|
A actual classificação das vasculites em idade pediátrica, validada em 2010 após período anterior em que era feita extrapolação a partir da dos adultos, consta do Quadro 3.
QUADRO 3 – Classificação das vasculites na idade pediátrica
| Adaptado de Ozen et al, 2006 |
I – Predominantemente de vasos de grande calibre
|
II – Predominantemente de vasos de médio calibre
|
III – Predominantemente de vasos de pequeno calibre A. Granulomatosas
B. Não Granulomatosas
|
IV – Outras vasculites
|
Seguidamente são abordadas as vasculites com maior relevância na idade pediátrica, salientando-se que a doença de Kawasaki (DK) integra capítulo próprio na Parte sobre Cardiologia.
1. PÚRPURA de HENOCH-SCHÖNLEIN
Definição, aspectos epidemiológicos e importância do problema
A púrpura de Henoch-Schönlein (PHS), também designada púrpura alérgica ou anafilactóide, é uma vasculite de pequenos vasos (capilares, arteríolas e vénulas), caracterizada por infiltração leucocitária na parede dos vasos sanguíneos, do que resulta aumento da permeabilidade vascular com hemorragia, edema e isquémia teciduais consequentes.
Trata-se da vasculite mais comum da idade pediátrica. A incidência anual oscila entre cerca de 9 e 18/100.000 crianças e adolescentes. É mais frequente no sexo masculino (1,5/1), sobretudo entre os 2 e 7 anos de idade (média de idade de 6 anos). Não existindo predilecção por raça, surge sobretudo no inverno e primavera. O aparecimento de casos familiares sugere componente genético. Os alelos HLA-B34 e HLA-DRB1*01 estão associados à nefrite de PHS.
Etiopatogénese
A etiopatogénese não está completamente esclarecida; admite-se o papel importante da hipersensibilidade a bactérias ou vírus (hipersensibilidade do tipo 3). Com efeito, a PHS surge geralmente após infecção do tracto respiratório superior (em cerca de 80% dos casos), sendo o Streptococcus b-hemoliticus do grupo A o agente mais frequentemente implicado. São também apontados como desencadeantes de PHS, infecções víricas, (Parvovírus, Adenovírus) ingestão de medicamentos (penicilina, ampicilina, eritromicina, quinino), alergénios alimentares e picadas de insectos.
Foi comprovada reacção antigénio-anticorpo no endotélio vascular levando a alteração da membrana capilar e a tendência hemorrágica. O mecanismo mais provável de indução da vasculite, com a intervenção do factor de necrose tumoral (TNF-a) e interleucina 6 (IL-6), é o depósito local de imunocomplexos (sobretudo IgA) nas paredes dos pequenos vasos sanguíneos da pele, glomérulos renais e mucosa gastrintestinal; o papel da activação do sistema do complemento é controverso. Não existe predisposição familiar, mas pode haver associação a deficiência congénita das fracções C2 e C4 b do complemento.
Como consequência deste processo verifica-se, através do exame histológico, a presença de infiltrados de leucócitos polimorfonucleares e mononucleares. Observam-se também trombos plaquetários, hemorragia e edema endotelial e, eventualmente, evolução para necrose fibrinóide de pequenos vasos sanguíneos.
Manifestações clínicas
O início da doença pode ser súbito ou gradual com aparecimento progressivo das respectivas manifestações. Em cerca de 50% dos doentes surge febre e mal-estar associados a infecção das vias aéreas superiores.
Sinais cutâneos
A pele é afectada em todos os casos (púrpura). A hemorragia cutânea manifesta-se inicialmente como exantema maculopapular ou urticariforme, raramente pruriginoso, indolor, na face dorsal das pernas junto à região maleolar dos tornozelos.
Ao cabo de 24-48 horas, as lesões tornam-se maiores, confluentes, purpúricas, não desaparecendo à compressão digital – com áreas concomitantes de petéquias confluentes e de equimoses de apresentação simétrica, principalmente na superfície de extensão dos membros inferiores e nádegas, menos frequentemente nos membros superiores e face, e raramente no tronco.
No mesmo doente poderão ser observados vários estádios evolutivos do exantema, sendo que as lesões mais antigas adquirem uma tonalidade mais escura, acastanhada, que tende a desaparecer em 4 semanas; no entanto, tais lesões poderão persistir durante meses (Figuras 1 A e B, e 2).
Em casos graves aparecem lesões necróticas e infecção secundária. Em cerca de 30-50% dos casos, principalmente em crianças com menos de 3 anos, ocorre edema não depressível de mãos, pés, região periorbitária, coiro cabeludo e, eventualmente, angioedema.
Poderá verificar-se uma ou mais recorrências (5-40% dos doentes) durante os primeiros meses de doença. Em casos de recorrência, as lesões tendem a aparecer nos mesmos locais das lesões prévias. O aparecimento de cicatrizes cutâneas é raro.
Sinais articulares
O compromisso articular (artrite e/ou artralgia) – que pode preceder as lesões cutâneas – constitui a segunda manifestação mais frequente, ocorrendo em 60 a 90% dos doentes. É geralmente agudo, migratório, oligoarticular, ao nível das grandes articulações, principalmente dos membros inferiores (joelhos, tornozelos), com duração de alguns dias em cada articulação. A artrite não deixa sequelas; porém, na fase aguda pode ser bastante dolorosa e incapacitante, de modo semelhante ao compromisso articular que surge na febre reumática. Poderá verificar-se raramente o aparecimento de mialgias associadas.
Sinais gastrintestinais
Em cerca de 30 a 70% dos casos ocorrem manifestações gastrintestinais no primeiro mês após o início da doença. Elas traduzem-se por dor abdominal periumbilical (tipo cólica de forte intensidade, exacerbada durante as refeições), náuseas, vómitos, hematemese, melena ou enterorragia; e, menos frequentemente, pancreatite aguda hemorrágica, colite ulcerosa, colite pseudomembranosa, enteropatia com perda de proteína, hidropisia da vesícula biliar e esteatorreia. A hemorragia gastrintestinal não ocorre na ausência de dor abdominal.
Complicações como invaginação intestinal, perfuração com peritonite, enfarte da parede intestinal ou estenose ileal podem aparecer no decurso da doença: as regiões mais acometidas são jejuno e íleo.
A invaginação intestinal (de localização ileoileal e ileocólica respectivamente em 65% e 35% dos casos) ocorre em 1 a 5% dos pacientes, traduzindo-se por dor abdominal intensa e súbita, hemorragia, e massa abdominal palpável. A dor abdominal intensa e o edema escrotal podem preceder o quadro de púrpura em cerca de 20% dos casos levando a confusão diagnóstica e, em muitos casos, a intervenção cirúrgica desnecessária; salienta-se que existe um risco maior de doença renal nos pacientes com hemorragia intestinal. Outra manifestação rara e tardia é a estenose esofágica.
O diagnóstico das situações que estão na base dos sinais gastrintestinais é realizado através de estudos radiológicos, ultrassonografia, endoscopia ou observação intra-operatória das lesões intestinais.
As lesões mais encontradas na endoscopia são áreas de hemorragia, erosões gástricas e jejunais, duodenite e lesões múltiplas de cólon e recto. Os exames contrastados são contra-indicados. As alterações histopatológicas cutâneas com evidência de vasculite (nas amostras obtidas por biópsia) confirmam o diagnóstico de PHS.
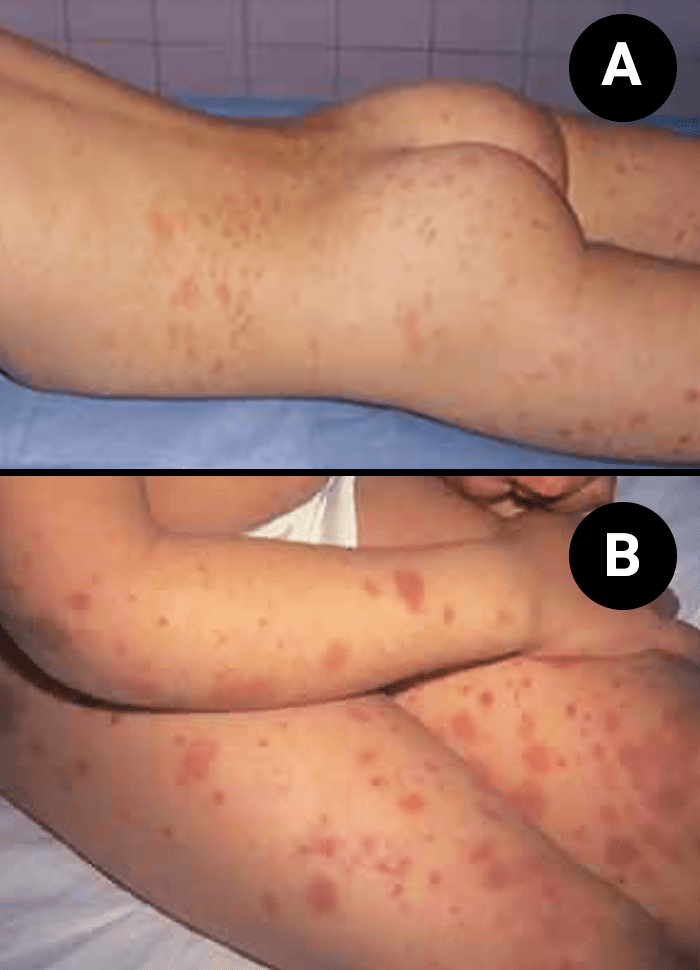
FIGURA 1. Púrpura de Henoch-Schönlein – Exantema maculopapular: A) localização nas nádegas; B) localização nas coxas e antebraços. (NIHDE)
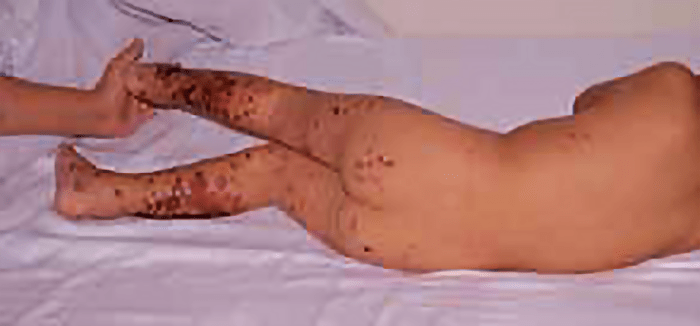
FIGURA 2. Púrpura de HS com localização de exantema predominante nas pernas
Sinais nefro-urológicos
De todos os sistemas orgânicos envolvidos, o rim é o principal responsável pela mortalidade, morbilidade e prognóstico a longo prazo. Com efeito, o compromisso renal está presente em 20 a 100% dos casos, sendo raro o seu início antes do quadro cutâneo. Em 80% dos pacientes esta manifestação surge nas primeiras 4 semanas de doença, e no restante contingente nos primeiros 2 meses, ou mais tarde. Verifica-se micro ou macro-hematúria em 25 a 30% dos pacientes. A proteinúria isolada é rara.
Hipertensão arterial, insuficiência renal aguda, síndromas nefrítica ou nefrótica são alguns dos determinantes de mau prognóstico. Pacientes com síndroma nefrítica e/ou nefrótica no início do quadro progridem para insuficiência renal crónica em 20% dos casos. A doença renal grave manifesta-se principalmente em crianças com idade superior a 5 anos e ocorre em 1 a 5% de todas as crianças com PHS. Factores como idade (crianças maiores), púrpura persistente, dor abdominal grave e recorrências, são apontados como preditivos do aparecimento e da gravidade de doença renal. A biópsia renal está indicada nos casos com síndroma nefrítica, síndroma nefrótica, insuficiência renal aguda ou crónica e proteinúria persistente. Em 10 a 20% dos casos a nefrite pode progredir, apesar da regressão das outras manifestações. Outras manifestações do foro nefro-urológico, mais raras, incluem: orquiepididimite com edema e dor escrotal, torção testicular, etc..
Outras manifestações associadas
Citam-se, neste contexto: febre, vasculite em sistema nervoso central (com cefaleias, convulsões, parésias, neuropatia periférica, hemorragia intracraniana, nevrite óptica, polirradiculonevrite, acidente vascular cerebral isquémico, alterações do comportamento ou coma), epistaxe, parotidite, compromisso ocular (hemorragia subconjuntival), hepatosplenomegalia, colecistite, tiroidite, linfadenopatia e alterações cardiopulmonares (enfarte do miocárdio, pleurite, pericardite, tamponamento cardíaco, doença pulmonar intersticial, edema ou hemorragia pulmonar, e diminuição da difusão pulmonar do monóxido de carbono, etc.).
A PHS pode acompanhar outras formas de vasculite ou de doença auto-imune, como por exemplo a febre mediterrânica familiar e a doença inflamatória do intestino.
Exames complementares
Os resultados dos exames laboratoriais são em geral inespecíficos.
A anemia, quando ocorre, é devida a perdas gastrintestinais. Leucocitose moderada com desvio à esquerda e eosinofilia são observadas nalguns pacientes. O número de plaquetas está geralmente normal, embora possa surgir hiperplaquetose relacionada com a gravidade da doença.
A velocidade de sedimentação (VS), a proteína C reactiva (PCR) e o complemento (C) podem apresentar-se normais ou discretamente elevados.
O coagulograma é normal. O antigénio relacionado com o factor de Von Willebrand encontra-se aumentado na fase activa da doença, assim como o tempo de tromboplastina parcial activado (TTPa), reflectindo lesão endotelial vascular. Os níveis de factor XIII, também conhecido como factor estabilizador da fibrina, podem estar diminuídos durante a fase aguda, principalmente nos casos com compromisso gastrintestinal grave. A fragilidade capilar poderá ser evidenciada pela prova do “garrote”.
O sedimento urinário está alterado nos casos de compromisso renal, mostrando hematúria com dismorfismo eritrocitário, com ou sem proteinúria, leucocitúria e, eventualmente, cilindros hemáticos.
A creatinina sérica pode estar elevada nos casos de insuficiência renal. A pesquisa de sangue oculto nas fezes pode ser positiva.
A imunoglobulina A sérica está aumentada em 50% dos casos; surge normalização ao cabo de 3 meses da doença, não havendo relação com o compromisso renal. A detecção de auto-anticorpos não tem valor na PHS.
O resultado da biópsia renal poderá evidenciar, através da técnica de imunofluorescência, deposição de imunocomplexos contendo IgA nos glomérulos renais, o que constitui factor importante no prognóstico. O quadro histopatológico é geralmente o de glomerulonefrite com lesões focais e/ou segmentares, culminando com a formação de imagens em “meia lua”.
A ecografia abdominoscrotal poderá ter interesse em situações acompanhadas de dor abdominal para confirmar ou infirmar diagnóstico de invaginação intestinal e/ou de torção testicular.
Diagnóstico
O diagnóstico da PHS é essencialmente clínico e é realizado quando a criança apresenta lesão purpúrica principalmente nos membros inferiores associada a artrite/artralgia, dor abdominal e hematúria/proteinúria.
Critérios de classificação para a PHS foram desenvolvidos pela European League Against Rheumatism (EULAR), Rheumatology International Trials Organisation (PRINTO) e Paediatric Rheumatology European Society (PRES) e ajudam na elucidação diagnóstica. (Quadro 4)
QUADRO 4 – Critérios de diagnóstico da Púrpura de Henoch Schönlein
| (Adaptado de Ozen et al, 2006) |
Púrpura palpável (critério obrigatório) na presença de, pelo menos, 1 dos seguintes achados:
|
No que respeita ao diagnóstico diferencial há que atender aos seguintes pontos:
- É importante afastar infecções graves como meningococcémia, septicémia e coagulação intravascular disseminada;
- O exantema hemorrágico, que ocorre ocasionalmente no sarampo, na varicela e na escarlatina, faz parte do diagnóstico diferencial da PHS;
- A vasculite de hipersensibilidade tem muitas semelhanças com a púrpura de Henoch-Schönlein. Em geral, a vasculite de hipersensibilidade provoca mais importante compromisso orgânico (pleurite, pericardite, insuficiência cardíaca), salientando-se que o predomínio de infiltrado de células mononucleares é frequentemente encontrado no exame anátomo-patológico. A ocorrência predominante em indivíduos maiores de 16 anos, característica da vasculite de hipersensibilidade, também diferencia as duas entidades. Outros tipos de vasculite e causas de glomerulonefrite e de diátese hemorrágica devem ser afastados;
- A síndroma hemolítica-urémica também pode simular alguns sinais e sintomas da PHS. A nefrite pós-estreptocócica pode ser diferenciada da púrpura pela diminuição dos níveis de complemento e ausência de sinais cutâneos;
- A nefropatia por IgA apresenta as mesmas alterações glomerulares detectadas por imunofluorescência e microscopia electrónica, embora com evolução clínica diferente da PHS. Refira-se que tal entidade (nefropatia por IgA) não apresenta o quadro cutâneo; por outro lado, evidencia sinais de lesão glomerular lentamente progressiva e crónica que pode levar à insuficiência renal crónica independente da apresentação, ao contrário da PHS que é uma doença aguda com lesão renal associada, geralmente não progressiva;
- Distúrbios da coagulação e trombocitopénia podem causar lesões purpúricas;
- Outros diagnósticos diferenciais nas situações acompanhadas de compromisso gastrintestinal incluem a doença inflamatória intestinal e a enterocolite por Yersinia;
- Os quadros de abdómen agudo aparente surgindo antes das lesões cutâneas podem ser confundidos com apendicite, adenite mesentérica, úlcera gastroduodenal, diverticulite e, eventualmente, levar à laparotomia exploradora;
- O chamado edema hemorrágico agudo, forma isolada de vasculite leucocitoclástica que afecta sobretudo crianças com < 2 anos, tem semelhanças com PHS. Verifica-se em tal situação bom estado geral com: febre, edema da face, escroto, mãos e pés; equimoses (maiores que na PHS) na face e extremidades; e petéquias nas mucosas. O tronco é poupado. Nos casos duvidosos pode recorrer-se à biópsia da pele.
Tratamento
Na maioria dos casos de PHS não existe necessidade de tratamento medicamentoso, uma vez que as manifestações são geralmente autolimitadas. Haverá, como medidas gerais, que providenciar a hidratação adequada. O tratamento específico, quando indicado, é de acordo com as manifestações clínicas apresentadas.
Em casos acompanhados de febre, edema ou sinais de compromisso articular, está indicado o uso de analgésicos-antipiréticos e de anti-inflamatórios. O ácido acetilsalicílico não deve ser usado pela possibilidade de provocar alterações gástricas e promover disfunção plaquetária. A ranitidina pode ser utilizada em pacientes com sintomas gastrintestinais, para redução da dor abdominal e da hemorragia digestiva.
O uso de glicocorticóides está indicado nos casos de lesão cutânea grave, com hemorragia, bolhas ou necrose; quadros gastrointestinais graves; orquite; comprometimento renal grave, com proteinúria intensa; e outras manifestações sisté
micas graves, como pancreatite, hemorragia pulmonar e vasculite do sistema nervoso central.
Nalguns casos de nefrite é necessário associar um imunossupressor, como azatioprina, ciclosporina, micofenolato ou ciclofosfamida. Na presença de glomerulonefrite rapidamente progressiva está indicada a plasmaférese. Os inibidores da enzima conversora de angiotensina são utilizados nos quadros renais com proteinúria.
Vale a pena ressaltar que o uso de glicocorticóides precocemente não previne o aparecimento do quadro renal e não devem ser utilizados para este fim.
Complicações e prognóstico
Geralmente a PHS regride ao cabo de 3-4 semanas, havendo, no entanto, a possibilidade de recorrências, como foi referido antes. A artrite também evolui favoravelmente, sem sequelas e sem recorrências.
No que respeita às manifestações do foro gastrintestinal há que contar com a possibilidade de hiperperistaltismo anómalo transitório com risco de invaginação intestinal e complicações inerentes, caso não seja feito diagnóstico atempado.
A mortalidade na fase aguda resulta de complicações gastrintestinais e de insuficiência renal aguda (rara).
A progressão para insuficiência renal crónica é inferior a 1% dos casos de PHS; no entanto, havendo antecedentes desta patologia na fase aguda, está indicado o seguimento da criança para avaliação periódica da função renal.
Em suma, o prognóstico é bom, em geral. Todas estas eventualidades deverão ser explicadas à família.
2. POLIARTERITE NODOSA
Definição, aspectos epidemiológicos e importância do problema
A poliarterite nodosa (PAN) é uma vasculite sistémica segmentar de tipo necrosante, atingindo sobretudo as artérias musculares de médio calibre, o que poderá levar a formação de aneurisma e estenose de artérias viscerais em qualquer órgão.
Os órgãos predominantemente afectados são: pele, vísceras abdominais, rins, sistema nervoso periférico e central e músculos.
Embora se trate de doença rara na infância e na adolescência, a PAN é a terceira vasculite mais frequente nesta faixa etária em alguns países, seguindo-se à PHS e à DK.
Apresenta dois subtipos: PAN cutânea e sistémica. A forma cutânea (limitada à pele) pode ser acompanhada de sintomatologia músculo-esquelética.
No grupo etário pediátrico verifica-se maior incidência entre aqueles de 7 a 11 anos, igualmente nos dois sexos; pode, no entanto, ocorrer em crianças muito pequenas. A sua distribuição é uniforme mundialmente, salientando-se maior número de casos descritos na Turquia e no Japão.
Etiopatogénese
A etiopatogénese da PAN é desconhecida, admitindo-se a hipótese de implicação de mecanismo imune ou de determinados agentes infecciosos (tais como Streptococcus dos grupos A e B, Parvovírus B19, Citomegalovírus, Vírus das hepatites B e C). Entre 10 e 50% dos casos associam-se ao vírus da hepatite B no adulto, contra cerca de 4% na idade pediátrica. Diversos estudos descreveram a associação da forma cutânea de PAN à infecção estreptocócica.
Recentemente foi descoberta a mutação com perda de função do gene CECR1, resultando na diminuição dos níveis da enzima adenosina deaminase 2 (ADA2), o que leva à vasculopatia e características clínicas semelhantes à da PAN.
A lesão arterial pode causar dilatação aneurismática (microaneurismas múltiplos de cerca de 1 mm) e nalguns vasos podem observar-se macroscopicamente nódulos; daí o termo de “nodosa”. Podem também ocorrer estenose, trombose e enfartes localizados. A necrose inicial é substituída posteriormente por tecido fibroso levando ao espessamento da adventícia, o que caracteriza a doença. Através da imunofluorescência é possível demonstrar depósitos vasculares de imunocomplexos (complemento e/ou imunoglobulinas).
Manifestações clínicas
O espectro de manifestações é muito variável, oscilando entre formas oligossintomáticas e graves e progressivas, por vezes fatais.
Nas formas mais frequentes, a doença tem início insidioso com sintomas inespecíficos como febre, perda de peso, astenia, mialgia e cefaleia.
Na PAN sistémica qualquer órgão interno pode estar comprometido, salientando-se que as áreas mais afectadas correspondem aos sistemas digestivo, renal e nervoso central (SNC) e periférico (SNP).
As manifestações clínicas mais frequentes integram as duas formas clínicas:
- PAN cutânea: sinais cutâneos, como livedo reticular, nódulos subcutâneos, necrose de extremidades, podendo haver mialgia e artralgia/artrite associadas;
- PAN sistémica, em que se verifica além do quadro cutâneo e músculo-esquelético, hipertensão, atingimento neurológico e gastrointestinal, podendo ainda afectar outros sistemas.
Globalmente as manifestações podem ser sistematizadas como se segue:
Sinais cutâneos
Nódulos subcutâneos (mais frequentes nos pés e face posterior das pernas, em geral no trajecto dos vasos sanguíneos); exantema máculo-papular, livedo reticularis, sinais de isquémia digital (edema, gangrena periférica, ulcerações, etc.); aos casos em que predominam os sinais cutâneos (PAN cutânea) podem estar associados outros sinais, adiante descritos (Figuras 3, 4 e 5).
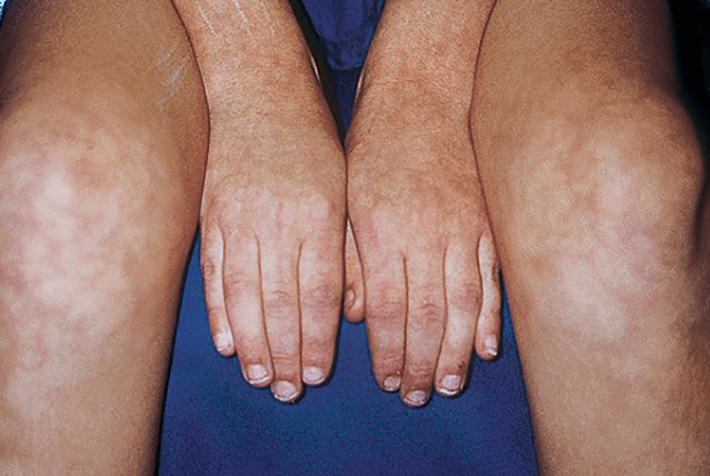
FIGURA 3 – PAN e livedo reticularis intenso em membros
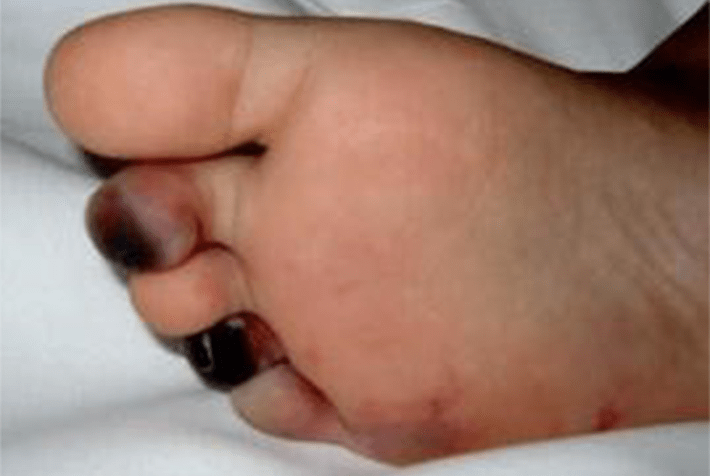
FIGURA 4 – PAN e necrose de 2° e 4° dedos do pé direito
Sinais músculo-esqueléticos
- Dor músculo-esquelética com fraqueza muscular gradual; artralgia, artrite (localizada ou difusa) e, por vezes, paniculite (inflamação do tecido adiposo subcutâneo).
Sinais abdominais
- Dor abdominal inespecífica relacionada com isquémia da artéria mesentérica ou outra artéria intra-abdominal; enfartes do intestino e pâncreas, assim como perfurações do tubo digestivo são também alterações possíveis.
Sinais renais
- Proteinúria, cilindrúria, síndroma nefrótica, hipertensão renovascular, insuficiência renal aguda, são alguns dos sinais de compromisso renal; a hipertensão arterial poderá, em certos casos, constituir uma manifestação isolada da doença.
Sinais neurológicos
- SNP: neuropatia periférica sensorial ou motora, sendo a manifestação característica a mononeurite múltipla, que evolui com “mão caída”ou “pé caído”.
- SNC: cefaleias, sinais focais, convulsões, hemiparésia, disfunção do cerebelo, paralisia de nervos cranianos e acidente vascular cerebral são manifestações raras. Os achados do SNC mais típicos são a presença de aneurismas e de micro-enfartes cerebrais múltiplos.
Sinais cardiovasculares
- Regurgitação das válvulas mitral ou tricúspide; diminuição da ejecção sistólica do ventrículo esquerdo, etc..
Sinais genitais
- Edema e dor testiculares, nódulos testiculares (raros).
Sinais oftalmológicos
- Episclerite
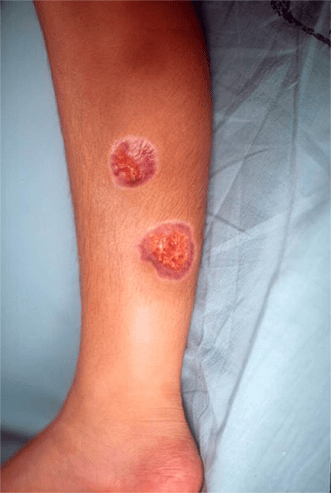
FIGURA 5 – Lesão ulcerada na região medial de membro inferior
Exames complementares
Face à anamnese e exame objectivo sugerindo vasculite com elevada probabilidade de PAN, são justificados determinados exames complementares (a solicitar criteriosamente e em função de cada caso particular), podendo afirmar-se que, de modo genérico, os respectivos resultados traduzem o carácter inflamatório da doença, sendo comum a alteração de parâmetros da fase aguda.
- Hemograma: anemia, leucocitose, trombocitose, etc.;
- VS elevada;
- PCR com valores elevados;
- Electroforese das proteínas séricas: hipergamaglobulinémia reflectindo activação policlonal das células B;
- Análise sumária da urina: proteinúria, hematúria;
- Doseamento da creatinina e ureia no sangue: elevação dos valores;
- Detecção do antigénio do factor VIII e da tromboglobulina: positividade reflectindo actividade da inflamação vascular – com interesse também na avaliação do resultado da terapêutica;
- Electrocardiograma: sinais possíveis de isquémia;
- Electromiograma: em caso de neuropatia periférica identifica os locais afectados;
- Angiografia/angiorressonância/angiotomografia: podendo demonstrar sinais de pequenos aneurismas mais frequentemente encontrados nas artérias mesentéricas, hepáticas e renais. Indicadas quando há compromisso de órgãos internos e, em função do contexto clínico de cada paciente, substituem eventualmente a biópsia;
- Biópsia da pele, incluindo nódulos subcutâneos, ou doutros órgãos: podendo identificar sinais de compromisso vascular (por ex. músculo, nervo, rim ou fígado: 1- inflamação destruindo a parede dos vasos, sendo característico o compromisso predominante das zonas de bifurcação; 2- presença de vasos com diversos estádios de lesão: alguns com sinais de inflamação aguda com infiltrado de neutrófilos, outros com infiltrado de mononucleares e, ainda outros e no mesmo território, com sinais de necrose e trombose, ao lado de vasos sem anomalias).
De realçar que o exame anátomo-patológico da região afectada ou a imagem angiográfica são fundamentais na confirmação diagnóstica da PAN, uma vez que os achados clínicos e laboratoriais são inespecíficos e podem ser semelhantes aos observados noutras vasculites e doenças reumáticas autoimunes.
Diagnóstico
O diagnóstico da PAN é feito através duma clínica compatível com a doença associada a biópsia ou angiografia características. Os critérios de classificação validados na infância pelos grupos internacionais European League Against Rheumatism (EULAR)/ Paediatric Rheumatology European Society (PRES)/ Paediatric Rheumatology International Trials Organisation (PRINTO) podem auxiliar no diagnóstico. (Quadro 5)
QUADRO 5 – Adaptação dos critérios de classificação da PAN na infância
| Critério obrigatório | Exame histopatológico com evidência de vasculite necrosante em pequenas e médias artérias. |
| 1. Envolvimento cutâneo | Livedo reticularis |
| 2. Sintomas músculo-esqueléticos | Mialgia ou miosite |
| 3. Hipertensão arterial sistémica | Pressão sistólica/ diastólica maior que o percentil 95 para altura |
| 4. Neuropatia periférica | Neuropatia periférica sensorial |
| 5. Envolvimento renal | Proteinúria > 0,3g/24h ou 30 mmol/mg de amostra isolada de albumina/creatinina |
| (Adaptado de Ozen et al, 2008) Nota – É necessária a presença do critério obrigatório (achado compatível na biópsia ou angiografia) associado a, pelo menos, mais 1 dos 5 critérios restantes. | |
O quadro clínico diversificado da PAN implica o diagnóstico diferencial com um conjunto de entidades clínicas, destacando-se:
- Lúpus eritematoso sistémico, dermatomiosite e esclerodermia, tendo em consideração que estas entidades podem cursar com lesões cutâneas vasculíticas, sendo que as restantes manifestações clínicas destas doenças permitem, em princípio, a distinção;
- Doença inflamatória do intestino ou neoplasias, tendo em consideração as manifestações do foro gastrintestinal que poderão acompanhar a PAN.
Tratamento
O tratamento da PAN inclui fundamentalmente:
- Corticoterapia: prednisolona PO, até máximo de 60 mg/dia; nos casos graves está indicado iniciar a corticoterapia com metilprednisolona EV em pulsos de 30 mg/kg/dia (até máximo de 1 g/dia) durante 3 dias, seguindo-se prednisolona PO com redução progressiva em função da resposta clínica e laboratorial do processo inflamatório;
- Imunossupressores em casos de falência da corticoterapia (persistência ou reactivação: ciclofosfamida PO, azatioprina PO, ou micofenolato; nas formas mais graves poderá estar indicado iniciar o tratamento com pulsos EV de ciclofosfamida (0,5-1 g/m2) segundo esquema que ultrapassa os objectivos do livro;
- Plasmaférese nos casos mais graves;
- Imunoglobulina EV, de utilização limitada, apenas em situações de falência da corticoterapia e antes de serem usados agentes citotóxicos;
- Terapia biológica para os casos graves e refractários, com terapia anti-TNF e anti-CD20 (rituximab);
- Profilaxia secundária de recorrência de infecção estreptocócica com penicilina benzatínica cada 21 dias nos casos de antecedentes de infecção estreptocócica prévia;
- Fisioterapia em função da sintomatologia músculo-esquelética;
- Regime alimentar adequado, com eventual suplemento de cálcio e vitamina D, evitando a iatrogenia.
Prognóstico
Com os recursos terapêuticos actualmente disponíveis e o diagnóstico precoce, o prognóstico desta doença tem melhorado, considerando-se dum modo geral bom. De referir, no entanto, que a possibilidade do compromisso dos órgãos atingidos e a iatrogenia, poderão agravar o prognóstico.
3. DOENÇA DE BEHÇET
Definição e aspectos epidemiológicos
A doença de Behçet (DB) é uma perturbação inflamatória sistémica que cursa com vasculite crónica e recorrente afectando grande variedade de vasos (vasculite variável primária), quer em calibre, quer em tipo – artérias e veias. Afecta principalmente a mucosa oral e genital, olhos e pele, mas o sistema músculo-esquelético, o gastrintestinal, o neurológico e o vascular também podem estar comprometidos.
Mais frequente na idade adulta (sobretudo segunda década) sem predomínio de sexo, é mais frequente nalgumas regiões do mundo, área mediterrânica, Japão e China (ao longo da Rota da Seda).
Na idade pediátrica atinge sobretudo pré-adolescentes e adolescentes (com uma incidência de cerca de 1/20.000 indivíduos; de salientar que se descreve hoje uma forma no recém-nascido e lactente.
Etiopatogénese
A classificação de doença autoinflamatória da DB é sugerida pela natureza episódica das manifestações, por se ter provado o papel preponderante da activação do sistema imune inato, pela ausência de autoanticorpos identificáveis, e pela associação com a febre Mediterrânica (MEFV).
Têm sido implicados, como prováveis desencadeantes, certos agentes infecciosos (vírus Herpes simples tipo 1, Parvovírus B 19, Streptococcus) induzindo respostas aberrantes do sistema imune inato. Por outro lado, diversos estudos têm demonstrado títulos elevados de anticorpos contra os respectivos agentes em determinados doentes e desencadeamento da doença pela exposição a organofosforados.
A distribuição geográfica, a frequência de aparecimento em familiares e a associação estatisticamente significante entre presença de antigénios do sistema HLA B5, B51, e doença de Behçet, sugerem predisposição genética.
A lesão patológica de base é vasculite das artérias de pequeno e médio calibre, com infiltração celular e consequentes necrose fibrinóide, estreitamento e obliteração dos lumes vasculares.
Manifestações clínicas
As manifestações clínicas são recorrentes com periodicidade muito variável. Por vezes o intervalo entre o primeiro episódio e o segundo pode ser de vários anos.
As úlceras orais são a manifestação mais frequente, ocorrendo em 50% a 80% dos casos. As úlceras são dolorosas, múltiplas, de 2 a10 mm de diâmetro, com centro necrótico e localizadas nos lábios, palato, língua, amígdalas, laringe e qualquer local do tubo digestivo. (Figura 6A)
Sendo a estomatite aftosa muito comum na primeira infância, as úlceras orais são consideradas como manifestação da doença de Behçet quando aparecem de forma recorrente (pelo menos 3 episódios por ano). Na maioria dos casos as úlceras regridem entre 7 e 14 dias, reaparecendo após períodos variáveis sem sequelas cicatriciais.
A presença de úlceras orais pode preceder durante anos uma segunda manifestação da doença, o que comporta dificuldade no diagnóstico.
As úlceras genitais, menos comuns na primeira infância, geralmente aparecem na puberdade; muito dolorosas e recorrentes, surgem no pénis (glande e prepúcio), escroto, vulva, vagina e região perianal com as mesmas características que as úlceras orais; contudo, esta localização deixa habitualmente cicatrizes.
O espectro de manifestações cutâneas é muito variado, incluindo: foliculite, púrpura, pústulas, vesículas, ulcerações, pápulas, furúnculos, abcessos, lesões acne-símile, eritema multiforme e eritema nodoso (este último em cerca de 50% dos casos).
As referidas lesões cutâneas desaparecem espontaneamente ao cabo de 7 a 14 dias. (Figuras 6-A, B e C)
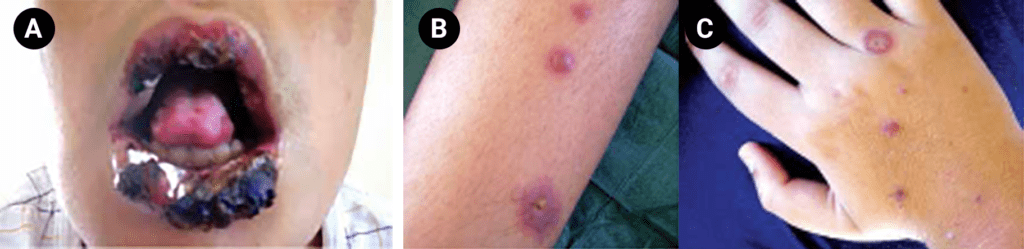
FIGURA 6. Jovem de 14 anos de idade, sexo masculino, com doença de Behçet diagnosticada aos 12 anos de idade. São evidentes úlceras orais e necróticas, estas últimas dificultando a abertura da boca (A); lesões papulopustulosas (acneiformes, pseudofoliculite) no antebraço (B) e na mão direita (C)
Uma manifestação cutânea considerada típica da doença – embora não patognomónica – é a chamada reacção patérgica (reacção do tipo pápula dura eritematosa superior a 2 mm, evoluindo para pústula estéril e úlcera em cerca de 2-3 dias) após picada com agulha.
As manifestações oculares, mais raras, são geralmente bilaterais caracterizando-se por alterações de segmento anterior (iridociclite) ou posterior (coriorretinite, papilite óptica, tromboflebite retiniana, arterite, etc.); também poderão surgir conjuntivite, episclerite e ceratite. O olho pode apresentar-se vermelho e doloroso. Poderão surgir complicações tardias como glaucoma, catarata e perda da visão.
As manifestações articulares, caracterizadas por oligo ou poliartrite ou artralgia, em geral das grandes articulações, não costumam deixar sequelas.
O compromisso vascular traduzindo-se por trombose de veias e artérias, corresponde à única vasculite que afecta os sistemas venoso e arterial. As manifestações mais comuns são a trombose de veias profundas (safenas, ilíacas, veia cava, veias dos membros superiores, supra-hepáticas – síndroma de Budd Chiari e cerebrais) e a tromboflebite superficial.
O compromisso arterial inclui aneurismas arteriais na aorta ou noutros vasos (inclusivamente artérias retiniana e pulmonar) com possível ruptura. A trombose arterial pulmonar constitui uma das manifestações associadas a mortalidade elevada.
A alteração mais grave da doença de Behçet é a neurológica, caracterizada principalmente por meningoencefalite asséptica, encefalomielite, hipertensão intracraniana secundária a trombose do seio dural, doença cerebelosa, hemiparésia, paralisia de pares cranianos, neuropatia periférica e alterações psiquiátricas. A cefaleia é um sintoma habitual.
As manifestações gastrintestinais podem ser inespecíficas (dor abdominal do tipo cólica, náuseas, vómitos e diarreia) ou semelhantes às verificadas nas doenças inflamatórias crónicas intestinais. Salienta-se o aparecimento de úlceras esofágicas e intestinais que causam dor e podem levar a quadros de melena e perfuração. Os quadros de estenose são mais frequentes do que os de perfuração, ao contrário do que acontece nos adultos. Por vezes existe hepato e/ou esplenomegália.
O Quadro 6 sintetiza os critérios de diagnóstico para a DB pediátrica.
QUADRO 6 – Critérios de classificação para a doença de Behçet (DB)*
| *A presença de 3 dos 6 critérios permite a classificação de DB na idade pediátrica. | |
| Sinais | Pontuação |
| Afta oral recorrente (pelo menos 3/ano) | 1 |
| Úlcera genital | 1 |
| Envolvimento cutâneo (foliculite, lesão acneiforme, eritema nodoso) | 1 |
| Envolvimento ocular (uveíte anterior, posterior ou vasculite retiniana) | 1 |
| Envolvimento neurológico (com excepção de cefaleia) | 1 |
| Trombose venosa, trombose arterial e aneurisma arterial | 1 |
Exames complementares
Determinados exames complementares têm utilidade para avaliar a actividade da doença, o diagnóstico diferencial e a exclusão de colagenoses. Eis os fundamentais:
- Hemograma: pode evidenciar anemia, discreta leucocitose e, raramente, neutropenia;
- Provas de actividade inflamatória como VS, PCR e alfa 2 globulina podem evidenciar valores aumentados na fase aguda da doença. As concentrações séricas de IgA e IgM encontram-se elevadas. O nível de complemento pode estar aumentado na fase aguda;
- Pesquisa de ANA e factor reumatóide: negativa;
- Pesquisa do antigénio de histocompatibilidade HLA-B51: positiva em cerca de 50 a 80% dos casos, segundo alguns estudos (associação frequente a compromisso ocular);
- Angiografia: importante para caracterizar as lesões vasculares;
- RM: importante para avaliar os efeitos da doença no sistema nervoso central.
Diagnóstico diferencial
Um vez que alguns doentes poderão não preencher os critérios da doença (doença de Behçet incompleta), a avaliar pela sensibilidade e especificidade dos critérios anteriormente descritos, doenças como lúpus eritematoso sistémico, artrite idiopática juvenil do tipo sistémico, doença de Reiter, doença inflamatória intestinal, eritema nodoso, estomatite aftosa e infecções herpéticas devem ser consideradas no diagnóstico diferencial.
Igualmente, a doença de Behçet deve fazer parte do diagnóstico diferencial doutras vasculites sistémicas, e de três outras entidades: síndroma de Stevens Johnson, eritema multiforme e necrólise epidérmica tóxica.
São referidas, a propósito, algumas características das três últimas:
1) A síndroma de Stevens Johnson integra quadro cutâneo vésico-bolhoso; inicia-se por máculas eritematosas com ulterior necrose central, formando-se consequentemente vesículas e bolhas que originam o aspecto de pele “desnudada” ou “descolada”, sobretudo na face, tronco e extremidades.
As lesões cutâneas, mais disseminadas que no eritema multiforme, são acompanhadas de lesões de duas ou mais mucosas, nomeadamente olhos, boca, via respiratória superior e tracto gastrintestinal, incluindo região anorrectal. Surge edema, eritema da mucosa bucal, bolhas e úlceras hemorrágicas. A referida síndroma está associada a infecções por Mycoplasma pneumoniae e certos fármacos como sulfonamidas, AINE, anticonvulsantes, antibióticos, etc..
2) O eritema multiforme consta fundamentalmente de lesões cutâneas de aspecto variável e aparecimento abrupto: máculas eritematosas, pápulas, vesículas, bolha ou lesões símile placas urticariformes confluentes ao nível da face, tronco e membros (rebordo eritematoso em coroa circular policíclica, centro pálido que evolui para arroxeado/necrótico). Quanto às mucosas apenas a boca é por vezes afectada: rubor dos lábios e boca. De etiopatogénese não esclarecida, está frequentemente associado a infecção por HSV.
3) A necrólise epidérmica tóxica, relacionável com os mesmos factores etiológicos da síndroma de Stevens Johnson, é considerada por alguns autores como uma forma mais grave de eritema multiforme. Relacionando-se com apoptose dos queratinócitos, traduz-se por áreas confluentes de eritema e “descolamento” da epiderme, associadas a inflamação das mucosas, das conjuntivas, boca e genitais (por vezes precedendo as lesões cutâneas). O sinal de Nikolsky é positivo: “descolamento” da pele após pressão tangencial da mesma.
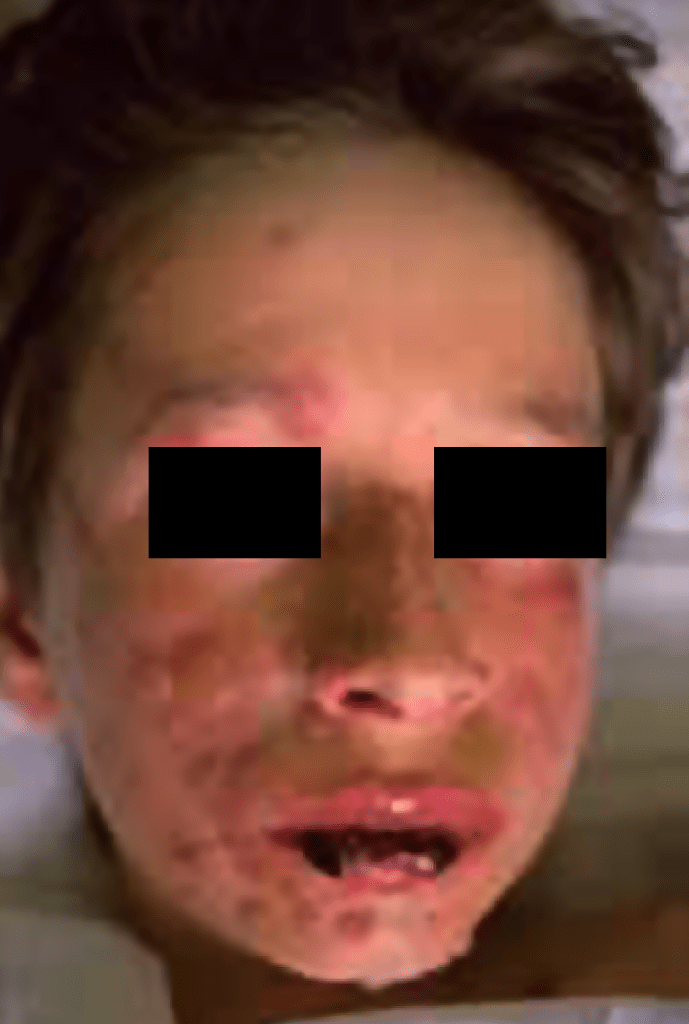
FIGURA 7. Síndroma de Stevens Johnson. (NIHDE)
Tratamento
Para além de aspectos gerais que incluem regime alimentar equilibrado e suplementos de cálcio e vitamina D, em função da idade, o tratamento da DB inclui determinadas medidas a seguir descritas:
- Para as úlceras orais extensas, úlceras genitais, manifestações cutâneas e articulares, a colchicina está indicada como primeira opção, e a penicilina benzatina pode ser associada nos quadros resistentes. Nos casos refractários, usa-se azatioprina e/ou glicocorticóides por curto período de tempo e, nos casos mais graves e resistentes, pode usar-se anti-TNF e apremilast (no contexto de úlceras orais e genitais). A talidomida é uma opção para o quadro mucocutâneo e os AINE para o quadro articular. Agentes tópicos, como glicocorticóides e sucralfato, podem ser usados nas úlceras orais;
- Para o comprometimento oftalmológico, as opções dependem da localização. Na uveíte anterior, colírios de glicocorticóide e midriáticos geralmente são suficientes para o controle da inflamação. Na uveíte posterior deve utilizar-se glicocorticóide sistémico e azatioprina. Nos casos de maior gravidade, associa-se a ciclosporina. Na doença refractária está indicado o uso de anti-TNF ou de interferon alfa 2a (IFN-α). No caso de utilização do IFN-α, a azatioprina deve ser suspensa;
- Para as manifestações intestinais está indicada a corticoterapia sistémica associada a sulfassalazina, azatioprina ou talidomida. E, nos casos graves, o anti-TNF;
- Para o comprometimento do SNC deve proceder-se a pulsoterapia de glicocorticóides, seguida por azatioprina. No caso de doença parenquimatosa refractária, os agentes anti-TNF e a pulsoterapia com ciclofosfamida são alternativa;
- Para o comprometimento vascular (trombose venosa profunda e trombose cerebral), utilizam-se glicocorticóides associados à azatioprina. Nos casos graves de trombose, como trombose de veia cava ou de supra-hepática, ou nos aneurismas, utiliza-se pulsoterapia com ciclofosfamida, seguindo-se terapia de manutenção com azatioprina. A anticoagulação, controversa, está contraindicada na presença de aneurismas de artérias pulmonares.
Prognóstico
Os casos fatais, raros (cerca de 3%), estão geralmente associados a perfuração gastrintestinal, doença neurológica e trombótica. Oclusões ou aneurismas de artérias são manifestações potencialmente fatais. De salientar que as sequelas oftalmológicas e as manifestações do sistema nervoso central são extremamente incapacitantes.
4. VASCULITES ASSOCIADAS A ANTICORPOS ANTICITOPLASMA DE NEUTRÓFILOS (ANCA)
As vasculites associadas ao ANCA (pANCA ou padrão perinuclear – ver atrás) são caracterizadas por: envolvimento dos pequenos vasos e presença de ANCA circulantes.
Actualmente considera-se que as vasculites associadas ao ANCA integram três formas distintas: granulomatose com poliangeíte/GPA (anteriormente designada granulomatose de Wegener), poliangeíte microscópica/PAM e granulomatose eosinofílica com poliangeíte/GEPA (anteriormente chamada Síndroma de Churg-Strauss).
Nesta alínea é dada ênfase às duas primeiras entidades.
4.1 GRANULOMATOSE com POLIANGEÍTE
Definição, aspectos epidemiológicos e importância do problema
A GPA é uma vasculite necrosante dos pequenos e médios vasos com formação de granulomas atingindo o tracto respiratório superior e inferior, e os rins (glomerulonefrite). Pode ocorrer de forma sistémica ou limitada, esta última podendo evoluir para sistémica.
Trata-se duma patologia rara na idade pediátrica, sobretudo antes dos 15 anos; na maioria dos casos surge entre a 4ª e a 6ª décadas de vida, sobretudo na raça caucasiana, e com predomínio no sexo feminino (~3/1). Estima-se uma prevalência entre 8 a 30 casos/1.000.000/ano, respectivamente no Reino Unido e nos Estados Unidos da América.
Etiopatogénese
A etiologia da GPA é desconhecida, admitindo-se que, devido à frequência do compromisso das vias aéreas, um antigénio inalado, (alergénio ou agente infeccioso), possa ser desencadeante dum processo inflamatório de mediação imune.
Algumas bactérias (nomeadamente Staphylococcus aureus) produzindo os chamados superantigénios (antigénios com capacidade para desencadear resposta inflamatória desproporcional ao estímulo), têm sido implicadas na iniciação do processo inflamatório ao nível dos capilares (capilarite).
A este poropósito, foi relatado o papel de um gene (CTLA-4 ou um gene próximo a este) codificando o antigénio 4 dos linfócitos T (linfócitos T citotóxicos) susceptíveis de maior activação no processo inflamatório.
Verificou-se que a proteinase-3 (PR3), em condições de normalidade integrada no interior dos grânulos – alfa dos neutrófilos, se encontra à superfície dos neutrófilos em doentes com GPA, sugerindo um papel etiopatogénico da expressão anormal de PR3 na doença.
O antigénio de histocompatibilidade HLA B8 parece estar associado à GPA; contudo outros haplótipos têm sido identificados em casos familiares de GPA associada a PAN (HLA A2, B7, DRw12, A31, Bw60 e DR4).
A marca histológica cardinal da afecção é a de vasculite necrosante, comum à PAM. Por biópsia renal demonstra-se a existência de sinais de glomerulonefrite com imagens de crescentes, e escassa (ou inexistente) deposição de complexos imunes (quadro pauci-imune), ao contrário do que se verifica em doentes com lúpus eritematoso sistémico. A biópsia evidencia inflamação granulomatosa, tal como na GEPA; porém não mostra infiltrados eosinofílicos perivasculares, achados característicos da última.
A razão pela qual o tracto respiratório e os rins constituem os alvos preferenciais na GPA é desconhecida.
Manifestações clínicas e diagnóstico
A sintomatologia respiratória está presente geralmente desde o início do quadro, podendo ser atribuída a infecções ou a alergia, principalmente quando estão ausentes as manifestações sistémicas. A persistência e/ou o agravamento dos sintomas, como rino-sinusite e otite média aguda recorrente, apesar da terapêutica para a alergia ou para a infecção, ou ainda o surgimento de complicações, como ulcerações da mucosa, perfuração do septo nasal ou deformidades nasais, levam à suspeita do diagnóstico.
As manifestações otorrinolaringológicas (rinite, otite, hipoacúsia, laringite, etc.) têm elevada prevalência (~70%), tanto no início do quadro clínico, como no decurso da doença. A obstrução laríngea e estenose subglótica surgem em geral como complicação tardia da GPA.
As manifestações pulmonares mais observadas são o infiltrado pulmonar e/ou nódulos, hemoptises, enfisema e pleurite.
O compromisso renal – que pode conduzir a insuficiência renal, surge em geral com a progressão da doença; traduz-se essencialmente por hematúria e proteinúria (na maioria dos casos). Hipertensão arterial e hematúria macroscópica são pouco frequentes.
Poderão surgir ainda alterações oculares (conjuntivite, esclerite ou proptose). Como manifestações menos específicas da GPA, citam-se febre, perda de peso, alterações cutâneas (púrpura palpável, úlceras, vesículas e nódulos subcutâneos), músculo-esqueléticas, e alterações do sistema nervoso (paralisia dos nervos cranianos, convulsões e neuropatia periférica).
Lesões papulares e nodulares do tipo acneiforme na face e membros superiores podem corresponder à manifestação inicial da doença em adolescentes.
O Quadro 7, adaptado de Ozen S, et al (EULAR/PRINTO/PRES, 2010), discrimina os critérios de classificação da doença (obrigatoridade de presença de, pelo menos, 3 em 6).
QUADRO 7 – Critérios de classificação da Granulomatose com Poliangeíte (GPA) em idade pediátrica
|
O diagnóstico deve ser suspeitado em crianças com sinusite grave associada a padrão radiográfico do tórax evidenciando sinais de granulomas/padrão de “infiltrado”, ou com quadro de glomerulonefrite. A TAC de alta resolução pode evidenciar outros sinais sugestivos como imagens de compromisso intersticial ou hemorragia pulmonar.
Diagnóstico diferencial
Os ANCA estão ausentes noutras doenças granulomatosas como sarcoidose ou tuberculose. A GEPA, vasculite que pode originar rino-sinusite crónica, distingue-se fundamentalmente da GPA pelos antecedentes de asma, eosinofilia no sangue periférico circulante e vasculite; por outro lado, na GEPA não se verificam lesões destrutivas na via aérea superior (Quadro 8). Noutras vasculites não se identificam os característicos granulomas ao nível de diversos órgãos, identificados na GPA por biópsia.
QUADRO 8 – Critérios de classificação da Granulomatose Eosinofílica com Poliangeíte/GEPA
| (sensibilidade de 85% e especificidade de 99,7%); Ozen et al, 2006 |
Verificação obrigatória de, pelo menos, 4 dos 7 critérios:
|
Exames complementares
Em cerca de 75% dos casos de GPA verifica-se anemia, elevação da VS, da PCR e das plaquetas. Os anticorpos da classe IgG c-ANCA dirigidos contra PR3 e identificados por imunofluorescência (padrão citoplasmático) são característicos da doença, porém a imunofluorescência também pode identificar padrão perinuclear em uma menor percentagem dos pacientes (p-ANCA).
Tratamento
Não há evidências científicas para o tratamento da GPA na faixa etária pediátrica; não provado cientificamente, é baseado em estudos em adultos.
A terapia de indução da remissão é feita com glicocorticóide associado à ciclofosfamida ou ao rituximab nos casos mais graves, e ao metotrexato nos casos leves de GPA localizada ao tracto respiratório. O rituximab é mais frequentemente utilizado nos casos refractários à ciclofosfamida e nas recidivas; porém está a ser cada vez mais utilizado como terapia de primeira escolha, considerando a sua menor toxicidade.
A plasmaférese é reservada para os casos de hemorragia alveolar difusa e glomerulonefrite rapidamente progressiva. Na terapia de manutenção da remissão usa-se azatioprina, metotrexato ou rituximab.
Prognóstico
Sem tratamento, a GPA é quase sempre letal (~ 82% de mortalidade no primeiro ano). O tratamento adequado e precoce permitiu melhorar o prognóstico, sendo este mais reservado em situações de hemorragia pulmonar e de insuficiência renal.
4.2 POLIANGEÍTE MICROSCÓPICA
Definição, aspectos epidemiológicos e importância do problema
A PAM é uma vasculite necrosante, predominantemente dos pequenos vasos, com características semelhantes às da GPA. Trata-se também duma patologia rara na idade pediátrica, sem predilecção de género. Entre as três vasculites antes referidas, é a forma mais grave.
Etiopatogénese
Em complemento do que foi referido a propósito da GPA, e das características histopatológicas distintivas dos três tipos de vasculite associadas a ANCA, cabe reforçar a ideia de que a respectiva etiopatogénese continua desconhecida, embora se tenha demonstrado o envolvimento de monócitos, neutrófilos e células endoteliais no processo.
Neutrófilos e monócitos são activados:
- pelos ANCA, especialmente contra antigénios citoplásmicos proteinase-3 (PR3) e mieloperoxidase (MPO); e
- pela libertação de citocinas proinflamatórias como TNF-alfa e IL-8.
Estas células inflamatórias, actuando sobre o endotélio, provocam a lesão vascular característica das vasculites associadas ao ANCA. Glomerulonefrite necrosante com presença de crescentes e capilarite pulmonar constituem dois achados histopatológicos relevantes. A razão pela qual o tracto respiratório e os rins constituem os alvos preferenciais na PAM é desconhecida.
Manifestações clínicas
As manifestações clínicas são semelhantes às da GPA, embora o quadro de afecção sinusal seja menos comum. Os sinais e sintomas de vasculite dos pequenos vasos incluem púrpura, artralgias, artrite e sinais gerais como febre, mal-estar, mialgias e perda de peso. A PAM afecta predominantemente o rim e pulmões; menos frequentemente outros sistemas são atingidos: SNC, pele, coração e globo ocular.
Exames complementares
Para além da elevação da VS e da PCR, o achado laboratorial típico é a presença de títulos elevados de ANCA, padrão perinuclear (p-ANCA, mieloperoxidase/MPO positiva por método ELISA).
Diagnóstico diferencial
Considerando as três vasculites necrosantes GPA, GEPA e PAM, para destinção, são utilizados os critérios:
- presença e teores séricos de ANCA-PR3 e ANCA-MPO; e
- verificação positiva ou negativa de inflamação granulomatosa.
Pode estabelecer-se o seguinte esquema:
| Nota: – Os anticorpos ANCA estão presentes em cerca de 90% de crianças com GPA (e em 70% dos casos com GEPA); – A presença de anti-PR3 aumenta a especificidade do teste. | |||
| Vasculite | GPA | GEPA | PAM |
| ANCA | PR3 | MPO > PR3 | MPO |
| Inflamação granulomatosa | + | + | – |
Tratamento
O tratamento da PAM na idade pediátrica também é baseado em estudos derivados da população adulta e segue o mesmo algoritmo feito para a GPA, levando em conta a gravidade de cada caso.
Importa realçar a importância do uso do trimetoprim-sulfametoxazol em pacientes com doença pulmonar para prevenção da infecção por Pneumocystis jiroveci, tanto na GPA quanto na PAM.
Prognóstico
A evolução é variável em função do grau de compromisso multiorgânico. Poderá haver recaídas em 60% dos casos. A evolução para insuficiência renal determina mau prognóstico.
5. ARTERITE DE TAKAYASU
Definição e importância do problema
A arterite de Takayasu (AT), também chamada doença sem pulso, é uma vasculite crónica progressiva de etiologia desconhecida que atinge grandes artérias, levando à formação de estenoses, dilatações e aneurismas.
Rara na idade pediátrica, afecta sobretudo o sexo feminino (4-10 F/ 1M), com uma incidência mundial entre 2,3 a 2,6 casos/1.000.000 habitantes/ano, sendo mais frequente em países asiáticos. A taxa de mortalidade é elevada (~40% na idade pediátrica).
Em casuísticas provenientes da Ásia, comprovou-se associação da doença a tuberculose.
Etiopatogénese
Na faixa etária pediátrica, a AT atinge principalmente a aorta e seus ramos primários, especialmente aorta abdominal e artérias renais.
Os achados imuno-histoquímicos sugerem um mecanismo de resposta imune a um antigénio, mediado por células T cujos receptores evidenciam disfunção. Por outro lado, demonstrou-se nalguns estudos a formação de imunocomplexos, a activação do complemento e o papel de anticorpos antiendoteliais. Verifica-se elevação de IL-6 e diminuição de IL-10.
Os achados histopatológicos evidenciam infiltrado linfo-monocitário, alterações granulomatosas e ulterior fibrose; este processo desenvolve-se da adventícia para a íntima.
No que respeita a antigénios de histocompatibilidade, verifica-se associação da doença ao HLA B-52 e HLA DR-2 no Japão, ao HLA B-52 e HLA B-5 na Coreia, e ao HLA B-39 no México.
Manifestações clínicas e diagnóstico
Numa fase precoce, com queixas inespecíficas de mal-estar geral, são notórios suores nocturnos, anorexia, perda de peso, fadiga, mialgia, artrite, seguindo-se por vezes hipertensão inexplicada.
A fase subsequente relaciona-se com sinais e sintomas de insuficiência vascular por múltiplas estenoses/oclusões arteriais. Essas manifestações clínicas dependerão da localização da artéria afetada, podendo ocorrer: acidente vascular encefálico, amaurose, dor torácica, dor abdominal, hipertensão e claudicação de extremidades.
Na idade pediátrica, relativamente ao adulto, é:
- mais evidente a diminuição ou ausência de pulsos periféricos; e
- menos evidente a claudicação da marcha por isquémia dos membros inferiores.
A presença do quadro descrito, assim como a verificação de VS e de PCR elevadas, obrigarão à auscultação periódica das grandes artérias, à palpação dos pulsos periféricos e à medição da pressão arterial nos quatro membros. (Quadro 9)
QUADRO 9 – Critérios de classificação da arterite de Takayasu
| Ozen e col., 2010 |
Presença de alterações angiográficas (critério obrigatório) e de, pelo menos, um dos cinco critérios:
|
A complicação mais temível da doença é a ruptura de aneurisma.
Os exames laboratoriais não são específicos e traduzem apenas o carácter inflamatório da doença. Assim pode-se observar anemia, leucocitose com neutrofilia e trombocitose; e elevação das provas de inflamação da fase aguda (VS e PCR). Poderá também verificar-se positividade do teste tuberculínico.
- O electrocardiograma permite evidenciar sinais de hipertrofia ventricular esquerda.
Os exames de imagem da árvore arterial são essenciais para o diagnóstico da AT e para o seguimento dos pacientes:
- A arteriografia, considerada “de padrão-ouro”, para delinear a alteração luminal do vaso;
- A angiotomografia (aTC) e angiorressonância (aRM), para avaliar a alteração luminal com a mesma precisão da arteriografia, assim como a parede arterial;
- A ultrassonografia com Doppler, para avaliação dos vasos cervicais, como artérias carótidas e vertebrais (Figura 8);
- A radiografia do tórax para detectar cardiomegalia ou contorno irregular do arco aórtico ou da aorta descendente.
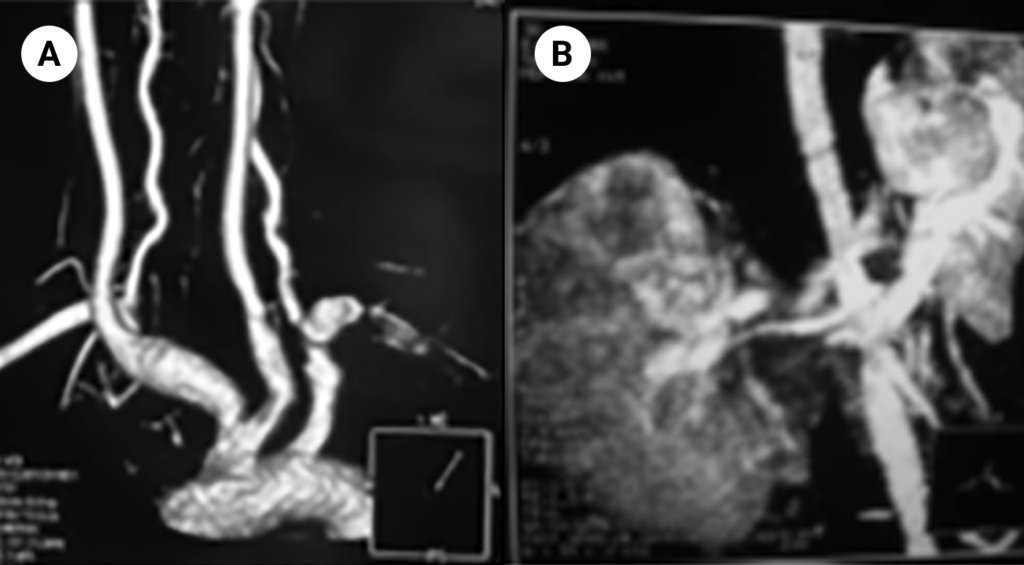
FIGURA 8. Imagens de angio-RM em criança de 9 anos de idade, com arterite de Takayasu diagnosticada aos 7 anos. Nestas imagens podem ser observadas várias estenoses arteriais, designadamente da artéria subclávia esquerda (A), da aorta abdominal e das artérias renais (B)
Diagnóstico diferencial
O diagnóstico diferencial da AT na idade pediátrica inclui um amplo espectro de situações, designadamente:
- Doenças que cursam com sintomas constitucionais prolongados, como tuberculose, neoplasias e algumas infecções víricas;
- Doenças que acarretam alteração estrutural da aorta ou de seus grandes ramos, tais como coarctação de aorta, displasia fibromuscular e doenças infecciosas;
- Doenças hereditárias, tais como neurofibromatose tipo I, síndromas de Ehlers Danlos tipo IV, e de Marfan; e
- Outras doenças inflamatórias, como febre reumática, doença de Kawasaki, espondilite anquilosante, de Behçet e lúpus eritematoso sistémico.
Tratamento
Os princípios básicos do tratamento da AT incluem medicação imunossupressora, com glicocorticóides, drogas sintéticas modificadoras do curso da doença (metotrexato é o mais utilizado na pediatria), ciclofosfamida e medicações imunobiológicas, como anti-TNF (adalimumab e infliximab) e anti-IL-6 (tocilizumab).
As estratégias terapêuticas dependem da gravidade e da refractariedade da doença em causa.
Em função do contexto clínico de cada caso, poderão também ser utilizados antiagregantes plaquetários, considerando o estado de hipercoagulabilidade da doença, anti-hipertensores, cardiotónicos e anticoagulantes.
Nos casos de tuberculose associada, há que proceder ao respectivo tratamento antes do tratamento específico da AT.
O tratamento cirúrgico, através de intervenção endovascular ou cirurgia aberta, está indicado nos casos de insuficiência valvar, estenoses significativas com lesão do órgão irrigado e hipertensão renovascular, entre outros
BIBLIOGRAFIA
Alfredo CS, Terreri MT, Hilário MO, et al. Púrpura de Henoch-Schönlein: recorrência e cronicidade. J Pediatr (Rio J) 2007; 83: 177-180
Andrews J, Mason JC. Takayasu’s arteritis: recent advances in imaging offer promise Rheumatology (Oxford) 2007; 46: 6-15.
Aries PM, Hellmich B, Voswinkel J, et al. Lack of efficacy of rituximab in Wegener’s granulomatosis with refractory granulomatous manifestations. BMJ 2006; 65: 853-858
Cabral DA, Canter DL, Muscal E. Comparing presenting clinical features in 48 children with microscopic polyangiitis to 183 children who have granulomatosis with polyangiitis (Wegener’s): An ARChiVe Cohort Study. Arthritis Rheumatol 2016; 68: 2514-2526
Clemente G, Hilário MO, Terreri MT, et al. Brazilian multicenter study of 71 patients with juvenile-onset Takayasu’s arteritis: clinical and angiographic features. Rev Bras Reumatol Engl Ed 2016; 56: 145-151
Clemente G, Silva CA, Sacchetti SB, et al. Takayasu arteritis in childhood: misdiagnoses at disease onset and associated diseases. Rheumatol Int 2018; 38: 1089-1094
Della Rossa A, Tavoni A, Merlini G, et al. Two Takayasu patients successfully treated with infliximab: a potential disease modifying agent? Rheumatology 2005; 44: 1074-1075
Dillon MJ, Eleftheriou D, Paul A, et al. Medium-size-vessel vasculitis. Pediatr Nephrol 2010; 25: 1641-1652
Eleftheriou D, Dillon MJ, Tullus K, et al. Systemic polyarteritis nodosa in the young: a single-center experience over thirty-two years. Arthritis Rheum 2013; 65: 2476-2485.
Eleftheriou D, Brogan PA. Vasculitis in children. Paediatr Child Health 2017; 28; 57-62. Erden A, Batu ED, Sönmez HE, et al. Comparing polyarteritis nodosa in children and adults: a single center study. Int J Rheum Dis 2017; 20: 1016-1022
Falcini F, La Torre F, Vittadello F, et al. Clinical overview and outcome in a cohort of children with polyarteritis nodosa. Clin Exp Rheumatol 2014; 32 (Suppl. 82): S134-S137
Iudici M, Puechal X, Pagnoux C, et al. Childhood-onset systemic necrotizing vasculitides: long term data from the French Vasculitis Study Group registry. Arthritis Rheumatol 2015; 67: 1959-1965
Iudici M, Quartier P, Terrier B, et al. Childhood-onset granulomatosis with polyangiitis and microscopic polyangiitis: systematic review and meta-analysis. Orphanet J Rare Dis 2016; 11: 141
Jelusic M, Vikic-Topic M, Batinic D, et al. Polyarteritis nodosa in Croatian children: a retrospective study over the last 20 years. Rheumatol Int 2013; 33: 3087-3090
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Koné-Paut I, Shahram F, Darce-Bello M, et al. Consensus classification criteria for paediatric Behçet’s disease from a prospective observational cohort: PEDBD. Ann Rheum Dis 2016; 75: 958-964
Martins KS, Baptista R, Nentzinsky V, et al. Poliarterite nodosa cutânea na infância com gangrena digital e possível associação com o estreptococo beta-hemolítico do grupo A: relato de caso e revisão de literatura. Rev Bras Reumatol 2008; 48: 111-117
Ozen S, Bakkaloglu A, Dusunsel R, Soylemezoglu O, et al. Childhood vasculitides in Turkey: a nationwide survey. Clin Rheumatol 2007; 26: 196-200
Ozen S, Marks S, Brogan P, Groot N, Graeff N, Avcin T. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatology (Oxford) 2019 [Epub ahead of print]
Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, et al. Paediatric Rheumatology International Trials Organisation (PRINTO). The EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II. Final classification criteria. Ann Rheum Dis 2010; 69: 798-806
Schwartz RA, Nervi SJ. Erythema nodosum: a sign of systemic disease. Am Fam Physician 2007; 75: 420-425.
Siomou E, Tramma D, Bowen C, Milford D. ANCA-associated glomerulonephritis/systemic vasculitis in childhood: clinical features–outcome. Pediatr Nephrol 2012; 27: 1911-1920
Terreri MT, Clemente G. Developments in large and midsize vasculitis. Rheum Dis Clin North Am 2013; 39: 855-875
Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 2014; 370: 911-920