1. DERMATOMIOSITE JUVENIL
Definição, importância do problema e aspectos epidemiológicos
A dermatomiosite juvenil (DMJ) é uma doença multissistémica rara, de etiologia multifactorial, caracterizada fundamentalmente por inflamação, aguda ou crónica não supurativa dos pequenos vasos, principalmente do músculo esquelético, da pele e do tracto digestivo.
Trata-se duma vasculopatia sistémica originando predominantemente lesão do músculo esquelético; na prática clínica pediátrica corresponde à miopatia inflamatória mais comum. Está associada a alterações cutâneas na quase totalidade dos casos.
Como características distintivas relativamente à DMJ do adulto cabe referir: 1) a vasculite é frequente e de gravidade variável; 2) poderão ulteriormente surgir calcinose subcutânea ou calcificações difusas das massas musculares envolvidas.
A incidência anual da DMJ, de acordo com diversos estudos epidemiológicos, oscila entre 1 a 4/1.000.000 de crianças entre 1 e 16 anos de idade, com pico de incidência entre 10-14 anos. É mais frequente no sexo feminino (2/1); nalguns grupos étnicos, como os judeus asiáticos e as crianças de raça negra, parece existir maior probabilidade de aparecimento.
No cômputo geral das doenças do foro reumático é menos frequente que a febre reumática, a artrite idiopática juvenil e o lúpus eritematoso sistémico.
Etiopatogénese
A causa da DMJ é desconhecida. Admite-se como hipótese para a génese da doença a comparticipação de determinados factores ambientais actuando num indivíduo imunogeneticamente predisposto. São raros os casos familiares, mas
estudos diversos demonstraram que a doença é mais frequente em determinados indivíduos portadores de certos antigénios de histocompatibilidade (HLA) determinados por genes no cromossoma 6; por exemplo: HLAA1, HLA B8, HLA DR3 e HLA DQ* 0501, sendo este último antigénio mais frequente na raça caucasiana.
Entre os factores ambientais mais implicados citam-se: agentes infecciosos actuando cerca de 3 meses antes do início da doença (Coxsackievirus B, Parvovírus, ECHO vírus, VEB, CMV, Streptococcus, Mycoplasma, Borrelia, Toxoplasma), imunizações (rubéola, BCG), reacções alérgicas, exposição ao sol, determinados fármacos, etc..
Diversos estudos apontam a autoimunidade como base etiopatogénica da doença: activação de linfócitos B, T e do complemento, por determinados antigénios, com deposição de IgG e IgM e imunocomplexos, (ou seja, produtos de reacção antigénioanticorpo associados a compostos de complemento) nas células endoteliais dos vasos e nos músculos, originando um processo de vasculite. A favor de lesão endotelial induzida por imunocomplexos está a demonstração de níveis plasmáticos aumentados de C3, de factor VIII e de fibrinopéptido A. Como resultado deste processo inicial surge um conjunto de lesões anátomo-patológicas com expressão especialmente relevante ao nível do músculo estriado, dos vasos e da pele.
O exame histológico do músculo estriado evidencia fundamentalmente: sinais de atrofia, degenerescência e necrose das fibras musculares; concomitantemente existe processo de regeneração das referidas fibras; com a evolução, as áreas de necrose são substituídas por áreas de tecido conjuntivo e de tecido adiposo.
No que respeita ao exame histológico dos vasos sanguíneos verifica-se, (como consequência da deposição de imunocomplexos) um processo de vasculite necrosante com infiltrado perivascular de células mononucleadas nos capilares, arteríolas e vénulas do músculo estriado, do tracto gastrintestinal, pele, e do tecido celular subcutâneo. A vasculopatia sistémica conduz a coagulação intravascular, com oclusão e enfarte consequentes.
Ao nível da pele, como consequência das lesões endoteliais capilares, surge atrofia da epiderme, degenerescência das células basais e infiltrado linfocitário
na derme. Na fase de cicatrização verifica-se deposição de sais de cálcio (calcinose). Em todo o trajecto do tracto gastrintestinal o epifenómeno mais marcante da vasculopatia é a ulceração e perfuração.
Manifestações clínicas
A DMJ tem geralmente início insidioso e progressivo, com aparecimento de mialgias e fraqueza muscular; no entanto, em cerca de 30% dos casos pode desenvolver-se de forma aguda e com rápida evolução das manifestações clínicas.
No início da doença prevalecem os chamados sinais e sintomas constitucionais como febre, anorexia, adinamia, astenia, perda de peso.
Na fase de doença estabelecida as principais características clínicas dizem respeito às manifestações cutâneas e musculares, sendo que as cutâneas podem ou não preceder as musculares; no primeiro caso, tais manifestações poderão ser interpretadas como “alergia ou outra dermatose”.
Como manifestações musculares citam-se: mialgias, contracturas e atrofia muscular, fraqueza muscular progressiva, principalmente proximal (cinturas escapular e pélvica, e flexores do pescoço e dorso), com dificuldade de subir e descer escadas, levantar-se da cadeira, levantar-se do chão, etc.. É nítido o sinal de Gower: o doente, ao levantar-se do chão uma vez sentado, na tentativa de se endireitar, tem de apoiar progressivamente as mãos sobre as pernas e coxas, de baixo para cima, alternadamente dum lado e doutro, como que servindo de alavanca ou suporte.
Se os músculos das vias respiratórias superiores e faringe forem afectados, poderá haver dificuldade na deglutição e voz nasalada.
As manifestações mucocutâneas, especialmente representativas da DMJ, são as seguintes:
- Exantema heliotrópico simétrico violaceoeritematoso circundando as pálpebras e podendo estender-se até ao dorso do nariz, regiões malares, ombros e dorso (em xaile ou manta); o exantema malar assemelha-se ao do lúpus eritematoso (Figura 1);
- Pápulas ou sinal de Gottron: elevações avermelhadas, lisas ou descamativas evoluindo para zonas atróficas e despigmentadas; localizam-se na superfície de extensão interfalângica, metcarpofalângica, região periungueal, cotovelos, joelhos, coxas e tórax (Figura 2); nos casos de vasculite disseminada pode verificar-se fotossensibilidade, livedo reticularis, etc. (Figura 3);
- Calcinose sob a forma de placas ou nódulos superficiais, geralmente nas extremidades, podendo ulcerar e infectar secundariamente;
- Eritema e/ou telangiectasia e espessamento periungueal, edema subcutâneo, nódulos subcutâneos, púrpura, alopécia e lipodistrofia (perda de tecido adiposo subcutâneo) localizada ou generalizada;
- Na mucosa oral: enantema, gengivo-estomatite com ulceração e odinofagia, associados a anticorpos anti-RNAt sintetase.
Outras manifestações incluem artrite (em cerca de 10-15% dos casos, em geral das pequenas articulações), tenossinovite, hepatomegália, esplenomegália, vómitos, diarreia, hematemeses, melenas, alterações cardiovasculares (pericardite, miocardite, hipertensão arterial, fenómenos de Raynaud), doença pulmonar restritiva associada a fibrose relacionada com a presença de anticorpos, exantema inespecífico, linfadenopatia, retinopatia associada a atrofia óptica, nefropatia podendo evoluir para insuficiência renal, etc..
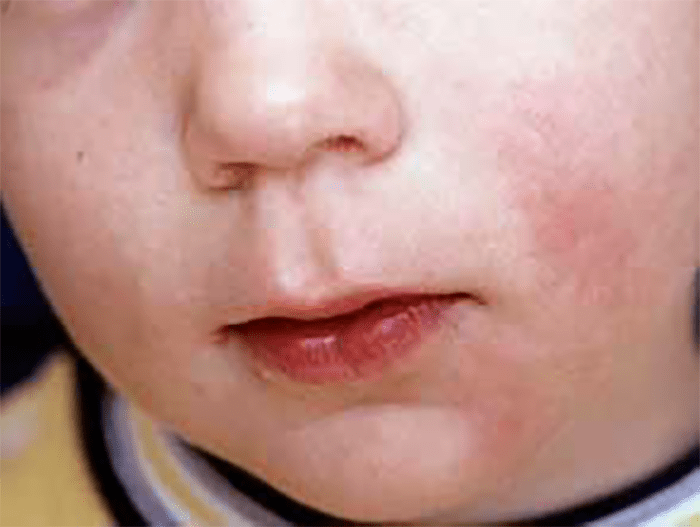
FIGURA 1. Exantema heliotrópico
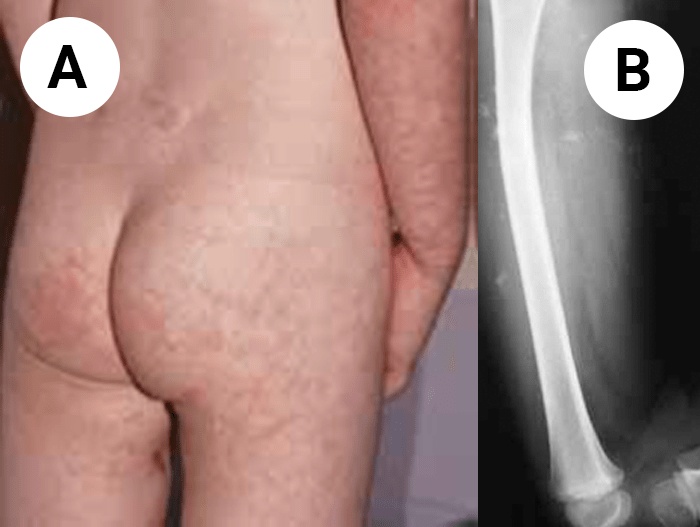
FIGURA 3. A) Caso de dermatomiosite em criança de 3 anos. São notórias as pápulas de Gottron no cotovelo e o livedo reticularis do antebraço e nádegas. B) A radiografia do braço mostra sinais de calcinose dos tecidos moles
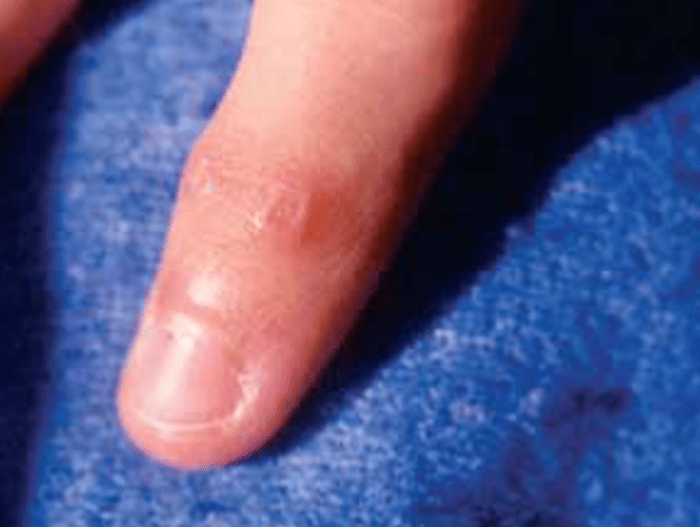
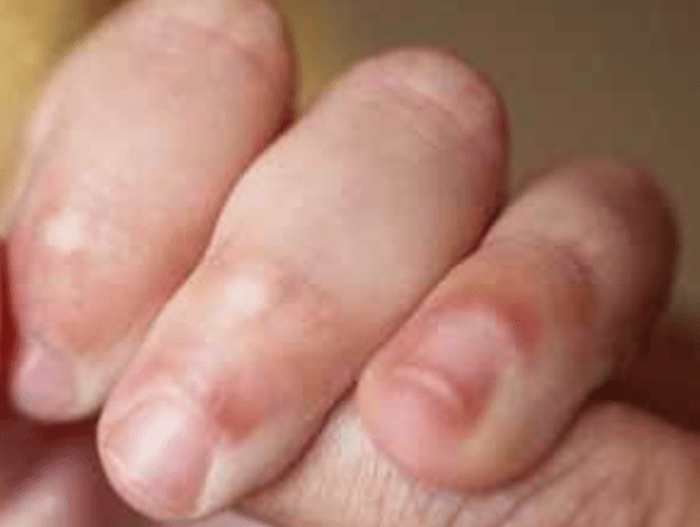
FIGURA 2. Pápulas de Gottron
Exames complementares
Face à suspeita de DMJ com base na anamnese e exame objectivo, certos exames complementares estão indicados para avaliar a repercussão do processo inflamatório em diversos territórios, nomeadamente ao nível do músculo. Os mais úteis são:
- Determinação de enzimas musculares, designadamente creatinofosfocinase (CK), desidrogenase láctica (LDH), aldolase, aminotransferases (ALT, AST), etc., as quais evidenciam valores elevados;
- Electromiografia (EMG), evidenciando anomalias do tipo fasciculações e descargas de alta frequência, padrão de miopatia e de desnervação, etc.;
- Biópsia muscular mostrando sinais de necrose e inflamação crónica.
Como critérios de diagnóstico de DMJ são considerados os seguintes parâmetros:
- Sinais cutâneos (exantema heliotrópico ou pápulas de Gottron ou telangiectasias ou espessamento periungueais);
- Fraqueza muscular proximal simétrica;
- Elevação dos valores das enzimas musculares;
- EMG evidenciando anomalias (padrão de miopatia e de desnervação);
- Biópsia muscular evidenciando anomalias (necrose e inflamação).
Assim, o diagnóstico definitivo de DMJ implica a observância de, pelo menos 4 critérios entre os 5 descritos, de -1) a -5). No entanto, desde que a clínica seja sugestiva, não deverá ser excluído o diagnóstico com menor número de critérios presentes.
Para além dos exames atrás descritos, outros poderão ser justificados em função do tipo de manifestações, ponderados caso a caso: hemograma (geralmente normal exceptuando nos casos de anemia por perda hemorrágica); VS e PCR (aumentadas somente em 50% dos casos); capilaroscopia (realizado com capilaroscópio ou oftalmoscópio de boa resolução) evidenciando por vezes sinais de dilatação capilar e de trombose; determinação do factor de von Willebrand que, como marcador de lesão endotelial, poderá indiciar actividade da doença ou recidiva; endoscopia digestiva; estudos imagiológicos (TAC, RMN,
etc.). Os indicadores da activação imunológica na DMJ incluem linfopénia, diminuição de: células circulantes memória CD8 ICAM-1, número absoluto de células natural killer (NT) CD56+, CD3–, CD16+ no sangue periférico.
Diagnóstico diferencial
Admitindo a possibilidade de formas clínicas raras de DMJ sem manifestações cutâneas, ou cujas manifestações cutâneas não constituem sinal precoce, o diagnóstico diferencial faz-se com situações acompanhadas de fraqueza muscular como miosite pós-infecções por vírus (influenza A e B, coxsackievirus), miopatias primárias, miosites inflamatórias associadas a doenças do tecido conectivo, polimiosite (causa importante de “bebé hipotónico”, mais rara que a DMJ), etc.. Em determinados casos a biópsia muscular permitirá fazer a destrinça.
Tratamento
As bases gerais da actuação na DMJ (salientando-se que deverá ser levada a cabo em centro especializado com o apoio de subespecialista) pressupõem:
- O facto de se tratar duma doença crónica com possibilidade de remissão ao cabo de 2 a 3 anos de evolução;
- O facto de que não existe tratamento curativo;
- A possibilidade de ser possível suprimir a resposta inflamatória e, até certo ponto, preservar a mobilidade articular e o crescimento e desenvolvimento adequados.
Para o tratamento da miopatia com sinais inflamatórios mínimos estão indicados os corticosteróides; em geral utiliza-se a prednisolona PO na dose de 1-2 mg/kg/dia; em função da resposta clínica e laboratorial e tendo em conta os efeitos secundários da corticoterapia de longa duração, procede-se depois à diminuição gradual da prednisolona para a 0,5 mg/kg/dia até suspensão.
Nos casos acompanhados de sinais inflamatórios acentuados, vasculite, de valor acentuado baixo (nº absoluto) de células CD56+ NH e de valor elevado das enzimas musculares, inicia-se a corticoterapia com doses mais elevadas (pulsos de metilprednisolona IV -30 mg/kg/dia durante 3 dias até máximo de 1 g/dia). Com a melhoria clínica e laboratorial reduz-se progressivamente a dose da metilprednisolona até 3, 2 ou 1 vez/semana, passando depois, a administrar prednisolona oral em dose de 0,5 mg/kg/dia nos dias de não pulsoterapia e após paragem da pulsoterapia.
A hidroxicloroquina na dose de 4 mg/kg/dia está indicada para as formas de manifestações cutâneas exuberantes, não tendo efeito na doença muscular.
O metrotrexato PO (15-20 mg/m2/semana acompanhado de suplemento de ácido fólico), outros imunossupressores (ciclosporina, ciclofosfamida ou azatioprina) ou IGIV, ou ainda agentes biológicos (etanercept) estão indicados nos casos refractários à corticoterapia (minoria).
Nos casos de miosite e manifestações sistémicas mais graves, deve ser utilizada terapêutica combinada, com corticosteróides, ciclosporina A (3 mg/Kg/dia) e metotrexato (15 mg/m2/semana).
Na fase aguda está indicado o repouso no leito; entretanto, o doente deve ser hospitalizado se houver sinais de vasculite, disfagia ou disfunção respiratória.
Uma vez que os raios solares provocam exacerbação dos sinais cutâneos, deverá providenciar-se o uso de protector solar.
Outras medidas incluem fisioterapia, cuidados cutâneos, suplementos de cálcio e vitamina D, regime alimentar adequado e protectores gástricos (estes últimos em situações com manifestações digestivas atrás descritas).
Complicações
A complicação mais grave, já descrita, é a calcinose da pele e tecidos moles, a qual pode ser generalizada.
Outra complicação descrita é a resistência à insulina evoluindo para diabetes do tipo 2, sendo de referir que o controlo de tal resistência é acompanhado de melhoria da doença muscular.
Prognóstico
A evolução da DMJ é variável: poderão surgir surtos de manifestações com duração de 6 a 8 meses (na maioria dos casos), de evolução benigna e seguidos de remissão total, ou surtos de duração superior a 2 anos, de evolução crónica, sem remissão, com sequelas (calcinose, atrofia muscular, contracturas, etc.) implicando corticoterapia prolongada.
Com a terapêutica actualmente disponível a mortalidade foi reduzida extraordinariamente (antes cerca de 40%, hoje cerca de 5%). As situações que comportam maior risco são: vasculopatia que afecta o tubo digestivo, provocando hemorragias e perfuração; miosite progressiva refractária à terapêutica e levando ao aparecimento ou ao agravamento da calcinose; infecções intercorrentes explicadas pelos efeitos da terapêutica imunossupressora; e, ainda, fraqueza muscular e insuficiência respiratória com risco de síndroma aspirativa.
2. POLIMIOSITE JUVENIL
Definições e sistematização
A polimiosite juvenil (PM) é uma miopatia inflamatória idiopática crónica muito rara, ainda menos comum que a dermatomiosite juvenil (DMJ), sendo que classicamente as referidas entidades clínicas se incluem no mesmo grupo; diferenciam-se apenas pela presença ou ausência de alterações cutâneas.
Trata-se de doenças musculares inflamatórias (miosites) crónicas que se traduzem clinicamente por diminuição da força muscular.
Outras miopatias inflamatórias incluem as miosites associadas às doenças do tecido conjuntivo e as pós-infecciosas.
Etiopatogénese
A etiopatogénese é sobreponível à da dermatomiosite juvenil; na PM não existem alterações cutâneas e o aparelho músculo-esquelético é acometido, em geral, de forma simétrica e proximal; por vezes existe localização focal e processo inflamatório mais ligeiro.
Manifestações clínicas
Apesar da raridade, a PM pode ser observada na primeira infância, constituindo uma das causas de “bebé hipotónico” por hipotonia generalizada.
Diagnóstico diferencial e exames Complementares
O diagnóstico diferencial da PM faz-se com outras miopatias e com perturbações neuromusculares não inflamatórias tais como:
- Distrofia muscular congénita;
- Distrofia muscular de Duchenne;
- Glicogenoses;
- Miopatias mitocondriais;
- Miopatias por drogas ou toxinas (D-penicilinamina, corticóides, hidroxicloroquina, etc.);
- Miopatias inflamatórias pós-infecciosas, mais frequentes em idade pediátrica (por vírus influenza A e B, coxsackievirus B, Mycoplasma, Salmonella, Serratia, Schistosoma, Toxoplasma, );
- Miosite eosinófila.
Tal como foi referido a propósito da DMJ, a biópsia muscular assume a maior importância.
Tratamento
Aplicam-se os mesmos princípios enunciados a propósito da DMJ. Quer nesta, quer na PM, quer ainda nas esclerodermias, em formas clínicas recorrentes ou resistentes à terapêutica convencional, alguns centros especializados têm aplicado com êxito imunoglobulina (IgG) por via subcutânea (sc/SCIG) em alternativa à IgG iv/IGIV, verificando-se menos efeitos adversos relativamente a esta última modalidade (IV).
BIBLIOGRAFIA
Aggarwal, Oddis CV. Therapeutic advances in myositis. Curr Opin Rheumatol 2012; 24: 635-641
Beaute J, Levy P, Millet V, et al.Economic evaluation of immunoglobulin replacement in patients with primary antibody deficiencies. Clinical and Experimental Immunology 2010; 160: 240-245
Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med 1975; 292: 344, 403
Cassidy JT, Petty RE (eds). Textbook of Pediatric Rheumatology. Philadelphia: Saunders, 2005
Castro TC, Yamashita E, Terreri MT, Len CA, Hilário MO. Calcinosis in juvenile dermatomyositis, a therapeutic challenge. Rev Bras Reumatol 2007; 47: 63-68
Cawkwell GMD. Inflammatory myositis in children including differential diagnosis. Currrent Opin Rheumatol 2000; 12: 430-434
Chari G, Laude TA. Juvenile dermatomyositis: a review. Int Pediatr 2000; 15: 21-25
Dalakas MC, Hohfeld R. Polymyositis and dermatomyositis. Lancet 2003; 362: 971-982
Fisler RE, Liang MG, Fuhlbrigge RC, Yalcindag A, Sundel RP. Aggressive management of juvenile dermatomyositis in improvide outcome and decreased incidence of calcinosis. J Am Acad Dermatol 2002; 47: 505-511
Goldman L, Schafer AI (eds). Goldman-Cecil-Medicine. Philadelphia: Elsevier Saunders, 2016
Haagenson L, Wong S, Brown KE. Parvovirus B19 and onset of juvenile dermatomyositis. JAMA 2005; 294: 2170-2171
Hoffman EP, Rao D, Pacham LM. Clarifying the boundaries between the inflammatory and dystrophic myopathies: insights from molecular diagnostic and microarrays. Reum Dis Clin North Am 2002; 28: 43-757
Hoffman F, Grimbacher B, Thiel J, et al. Home based subcutaneous immunoglobulin G replacement therapy under reallife conditions in children and adults with antibody deficiency. Eur J Med Research 2010; 15: 238-245
Jolles S, Stein MR, Longhurst HJ, et al. New frontiers in subcutaneous immunoglobulin treatment. Biologics in Therapy 2011; 1: 3-6
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Pachman LM, Khojah AM. Advances in juvenile dermatomyositis: myositis specific antibodies aid in understanding disease heterogeneity. J pediatr 2018; 195: 16-27
Reed AM. Myositis in children. Current Opin Rheumatol 2001; 13: 428-433
Romero KT, Terreri MT, Len CA, Hilário MO. Juvenile dermatomyositis e polymyositis: diagnostic and treatment. Rev Paul Pediatria 2003; 2: 223-227
Ruetter A, Luger TA. Efficacy and safety of intravenous immunoglobulin for immune-mediated skin disease. Am J Clin Dermatol 2004; 5: 153-160
Vehe RK, Riskalla MM. Collagen vascular diseases: SLE, Dermatomyositis, Scleroderma, and MCTD. Pediatr Rev 2018; 39: 501-513