Sistematização
O Quadro 1 sistematiza as principais situações clínicas relacionadas com alterações do conteúdo escrotal. A hérnia inguinoscrotal é abordada noutro capítulo.
QUADRO 1. Alterações do conteúdo escrotal
|
1. TESTÍCULO ECTÓPICO
Definições
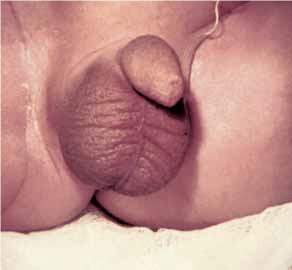
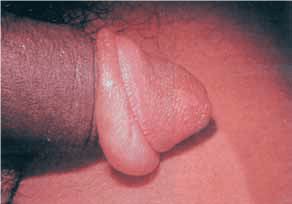
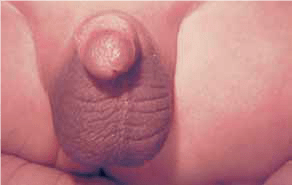
Testículo ectópico (ou distopia testicular) significa testículo em situação anormal (ectopia ou distopia: significa situação anormal de um órgão, em geral de origem congénita). Tal situação verifica-se como resultado de os testículos não ocuparem a sua posição normal intraescrotal.
O âmbito da distopia abrange diversas modalidades em função da etiopatogénese:
1 – Criptorquidia que corresponde à situação de testículo não descido (que não pode ser manipulado para a bolsa escrotal, encontrando-se localizado em algum ponto do seu trajecto normal de descida); pode estar localizado no interior da cavidade abdominal, ou no canal inguinal (na maioria das vezes). Com uma incidência de cerca de 2% a 6% nos recém-nascidos de termo e cerca de 30% nos pré-termo, a criptorquidia é unilateral em 80-90% dos casos. Pode estar associada a anomalias diversas.
2 – Testículo ectópico propriamente dito (raro) que corresponde à situação de testículo fora do trajecto normal de descida, geralmente na coxa ou no períneo, região suprapúbica ou bolsa escrotal contralateral. Também unilateral em 80-90% dos casos.
3 – Testículo retráctil que corresponde à chamada situação de testículo “em ascensor”; há mobilidade do testículo no trajecto do canal inguinal, entre a bolsa escrotal e o anel inguinal superficial. Uma vez “agarrado” pelos dedos do observador é levado facilmente à bolsa escrotal, aí permanecendo sem grande tensão. Na maior parte dos casos é bilateral, explicando-se a “subida como um ascensor” pela hiperactividade do músculo cremasteriano. Esta tendência deixa de ser notória com a idade por diminuição progressiva da hiperreflexia cremasteriana.
Etiopatogénese
A descida normal do testículo processa-se por volta do 7º mês de gestação. A não descida pode explicar-se por diversos factores:
- Alterações hormonais;
- Disgenésia do testículo;
- Anomalia anatómica.
Na primeira fase da descida testicular assumem importância o factor hormonal ILF3 (insulin-like factor 3) e o receptor LGR8 (leucin-rich repeat-containing G protein-coupled receptor 8).
As anomalias genéticas mais frequentemente associadas a criptorquidia são a síndroma de Klinefelter (47XXY ou 46XY / 47XXY) e as mutações no gene do receptor INSL3.
Quanto mais elevada a posição do testículo, maior é a probabilidade de disgenésia (anomalia de diferenciação sexual acompanhada de anomalia congénita) e, portanto, de infertilidade e de ulterior malignização.
Com efeito, como resultado da localização anómala, tornam-se evidentes alterações histológicas durante o segundo ano, as quais se caracterizam por insuficiência na transformação dos gonócitos em espermatogónias diferenciadas, com oligospermia, atrofia das células de Leydig e anomalias nas células de Sertoli. Têm sido descritas anomalias de desenvolvimento das células germinais no testículo contralateral nos casos unilaterais, o que sugere a possibilidade de um defeito de desenvolvimento global ou endócrino; tal é igualmente corroborado pela presença frequente de defeitos do epidídimo.
Quando se verifica criptorquidia isolada na data do nascimento, o testículo pode descer em 1/3 dos casos nos primeiros 6 meses de vida; e, se tal não acontecer até ao final do primeiro ano de vida, já não se verificará o processo de descida. De referir que o testículo pode estar localizado na bolsa na data do nascimento e subir depois.
A não descida do testículo é acompanhada de: hérnia inguinal em 90% dos casos (o que se torna lógico, pois o canal peritoneovaginal só se encerra depois de o testículo chegar ao fundo da bolsa escrotal); e de hipospadia em 10% dos casos.
Complicações
São descritas as seguintes complicações: torção, malignização (na terceira década de vida), atrofia, infertilidade (nos casos bilaterais) e psíquicas (em relação com problemas de estética anatómica).
Diagnóstico
A presença de hidrocele (ver adiante) no recém-nascido e a verificação de testículo retráctil entre os 6 meses e a puberdade, dificultam a identificação da posição do referido testículo.
Para o correcto diagnóstico torna-se importante salientar determinados gestos semiológicos.
A inspecção deve começar pela verificação da simetria das bolsas, bem como dos sulcos cutâneos transversais das mesmas. Quanto mais atrófica e aplanada for a bolsa, com os sulcos quase inexistentes, maior é a probabilidade de se estar em presença de um testículo atrófico intrabdominal, ou mesmo de ausência testicular.
Por outro lado, uma bolsa bem desenvolvida, com sulcos normais coexistindo com não palpação de testículo, deve levar a admitir a hipótese de ter sido habitada por um testículo que, entretanto, desapareceu gradualmente por um mecanismo de torção, com a consequente isquémia e atrofia; é o conceito de testículo evanescente.
A palpação deve ser feita em ambiente calmo, se necessário no banho, com aquecimento prévio das mãos e, na criança mais velha, em posição bípede e com a perna cruzada. Todas estas manobras têm como finalidade atenuar o reflexo cremasteriano e tentar diferenciar testículo não descido verdadeiro de testículo retráctil.
Se o testículo se palpar na região inguinal e não se conseguir colocar na bolsa ou, uma vez colocado nesta, ele subir de imediato, considera-se não descido. Se houver dúvidas deve repetir-se o exame noutra ocasião.
Na hipótese de o testículo não se palpar no canal inguinal, devem ser examinados possíveis locais de ectopia – púbis, coxas ou períneo, etc..
Exames complementares
Os estudos imagiológicos (ecografia) são pouco úteis para o diagnóstico, excepto em crianças obesas com testículos inguinais; são importantes, contudo, para o seguimento pós-operatório (dimensões e estrutura).
A ressonância magnética nuclear com injecção de gadolinium pode ser útil nalguns casos de ausência de identificação testicular quando se procede à exploração cirúrgica.
O método mais eficaz de localização (e tratamento) do testículo não palpável é a laparoscopia.
Tratamento
O tratamento engloba duas modalidades:
Hormonoterapia
Esta modalidade é controversa. A única indicação formal da terapêutica hormonal é a situação de testículo palpável bilateral retráctil alto. Utiliza-se gonadotrofina coriónica humana (HCG) em doses baixas – geralmente 9.000 UI, divididas em 6 doses parciais de 1.500 UI, por via intramuscular (2 vezes por semana, durante 3 semanas). Em doses pequenas os efeitos virilizantes não se verificam ou regridem facilmente.
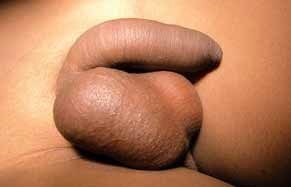
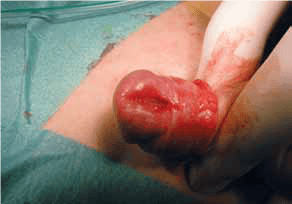
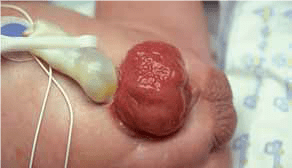
FIGURA 1. Torção testicular. A – Tumefação e edema do escroto;
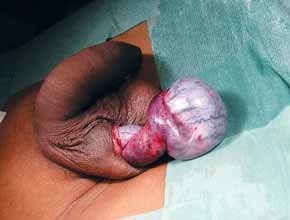
FIGURA 1. Torção testicular. B – Aspecto intra-operatório.
Há autores que preconizam HCG em pequenas doses em todos os casos de distopia, precedendo a intervenção cirúrgica no pressuposto de que tal terapêutica facilita a mesma.
Pode utilizar-se igualmente o factor libertador da hormona luteinizante (LHRH) por via intranasal durante 4 semanas.
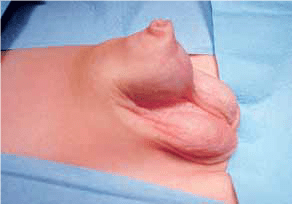
Intervenção cirúrgica
A idade ideal para a intervenção cirúrgica situa-se entre 1 e 2 anos, consistindo em orquido-pexia. Pode ser feita em regime ambulatório, sendo as complicações raras: atrofia e retracção. Os doentes devem ser seguidos anualmente devido às possíveis complicações, em especial risco de malignização nos testículos disgenésicos na terceira década de vida.
A cirurgia está ainda indicada para tratamento das criptorquidias iatrogénicas, isto é, secundárias herniorrafia inguinal anterior.
2. TUMEFACÇÃO DO ESCROTO E ESCROTO AGUDO
A tumefacção do escroto pode ser aguda ou crónica, dolorosa ou indolor. Como exemplos de tumefacções dolorosas citam-se: torção do testículo, torção do apêndice testicular, epididimite, lesão traumática/hematocele, hérnia inguinoscrotal encarcerada, orquite da papeira, etc..
Como exemplos de tumefacções acompanhadas de desconforto ou não dolorosas são referidos: tumor testicular, edema escrotal no contexto de púrpura vascular (PHS), varicocele, hidrocele, hérnia inguinoscrotal, etc.. (ver adiante)
As situações classicamente designadas por escroto agudo abrangem um conjunto de entidades discriminadas no Quadro 2 as quais pressupõem a necessidade de intervenção urgente ou emergente. No caso de torção do testículo (com confirmação diagnóstica) haverá necessidade de intervir no período que não ultrapasse seis horas a fim de garantir viabilidade do mesmo. (Figura 1)
As manifestações clínicas do escroto agudo são essencialmente dor do escroto surgida de modo súbito (na criança pequena poderá ser o choro ou irritabilidade que alertam) e edema.
Uma nota importante para o papel indispensável da ecografia-doppler no diagnóstico diferencial entre epididimite e torção do testículo. Na hipótese de tal exame complementar ser inexequível, a intervenção cirúrgica não deve ser diferida, sendo preferível uma intervenção “branca” a um diagnóstico de torção não feito.
QUADRO 2 – Escroto agudo
| Torção do testículo No lactente ou na puberdade: testículo e cordão espermático dolorosos à palpação, hiperestesia e maior dureza ao tacto; rubor do escroto; pode surgir no feto, o que conduz inevitavelmente a testículo inviável. |
| Torção do epidídimo Mais frequente entre os 4 e 8 anos: edema do escroto sem rubor aparente; aparentemente o pólo superior do testículo está mais sensível à palpação, verificando-se a este nível uma “mancha azulada” através da transiluminação. |
| Edema idiopático do escroto Também mais frequente pelos 4-8 anos, acompanhando-se de eritema que ultrapassa os limites do escroto; dum modo geral não é doloroso à palpação. |
| Epididimite Raramente surgindo antes da puberdade, o epidídimo está mais sensível ao tacto; esta situação é por vezes acompanhada de infecção urinária com refluxo. |
| Hérnia inguinal irredutível |
3. VARICOCELE
Trata-se de varicosidades das veias testiculares, mais frequentes no lado esquerdo, podendo ocorrer na puberdade.
Em casos especiais poderá estar indicada a obliteração cirúrgica da veia testicular, utilizando a via laparoscópica.
4. HIDROCELE
O hidrocelo (ou a hidrocele) é uma acumulação de líquido na tunica vaginalis, situação que surge em cerca de 1-2% de RN do sexo masculino. Na maior parte dos casos é “não comunicante” o que resulta de o processus vaginalis ter ficado obliterado durante o processo de desenvolvimento. Em tais casos desaparece por volta do 1 ano de idade.
Se o referido processus vaginalis continuar permeável, o hidrocelo persiste, sendo que as suas dimensões aumentam “por enchimento” em posição bípede e diminuem em posição de decúbito.
Uma das complicações do hidrocelo comunicante é o desenvolvimento de hérnia inguinal.
O exame físico evidencia escroto distendido, não sob tensão, e difusão da luz por transiluminação, o que traduz a existência de líquido.
Nos casos que persistem para além dos 18 meses está indicada intervenção cirúrgica.
5. QUISTO DO CORDÃO
Trata-se duma tumefacção quística esferóide no trajecto do cordão espermático mais frequentemente na região inguinal. Resulta da persistência do canal peritoneovaginal, que é permeável a conteúdo líquido, manifestando-se clinicamente por uma tumefacção da região inguinal, o que implica diagnóstico diferencial com as massas inguinais.
O quisto do cordão evidencia ao exame clínico uma consistência relacionável com conteúdo fluido, móvel, não redutível; frequentemente é possível determinar os seus limites: orifício inguinal interno, na sua porção proximal e o orifício inguinal superficial, na sua porção mais distal.
Esta particularidade permite distinguir esta entidade da hérnia inguinal, em que há permeabilidade do canal peritoneovaginal para conteúdo visceral, sem limites definidos, uma vez que a hérnia pode ocupar todo o canal e ter extensão intra escrotal e ser redutível.
O diagnóstico diferencial faz-se com as massas inguinais, em geral.
Embora o exame clínico cuidadoso permita chegar ao diagnóstico, cabe referir o papel do exame ecográfico com sensibilidade e especificidade elevadas.
O aumento de volume ou dor constituem indicação para intervenção cirúrgica; esta consiste na laqueação alta do canal peritoneovaginal e na plastia do orifício inguinal profundo, procedimento cirúrgico que é realizado em regime de ambulatório.
6. ESPERMATOCELE
A espermatocele consiste numa dilatação quística do epidídimo devida a acumulação de secreções espermáticas; trata-se, pois, dum quisto do epidí- dimo.
Clinicamente é diagnosticada como uma tumefacção elástica, associada ao epidídimo, indolor e sem características inflamatórias.
A terapêutica pode ser realizada por punção aspirativa ou excisão cirúrgica por abordagem transcrotal.
7. TUMOR TESTICULAR
Os tumores testiculares podem aparecer em qualquer idade. Na maior parte dos casos constituem massas indolores que não transiluminam. Em caso de suspeita deve proceder-se a ecografia. A alfa-feto-proteína e a beta-gonadotrofina humana coriónica estão elevadas respectivamente nos teratomas, e nos coriocarcinomas e germinomas.
Nos casos de malignidade está indicada a orquidectomia radical.
BIBLIOGRAFIA
Agarwal PK, Diaz M, Elder JS. Retractile testis – Is it really a normal variant? J Urol 2006; 175: 1469-1499
Ashcraft KW, Holder TM (eds). Paediatric Surgery. Philadelphia: Saunders, 2003
Barthold JS. Is adjuvant hormonal therapy indicated in cryp- torchidism. Nat Clin Pract Urol 2005; 2: 366-367
De Kretser DM. Differences in the prevalence of cryp- torchidism. Lancet 2004; 363: 1250-1252
Hutson JM, Hasthorpe S. Testicular descent and cryp- torchidism in 2004. Pediatr Surg 2005; 40: 297-302
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics.NewYork:Mc Graw Hill Education, 2018
McInerny T(ed). Tratado de Pediatria / American Academy of Pediatrics. Madrid: Panamericana, 2010
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Mouriquand P. The normal testis. Arch Dis Child 2007; 92: 3
O’Neil JrJA, Rowe MI, Grosfeld JL, et al (eds). Pediatric Surgery. Philadelphia:Elsevier, 2017
Sandlow J. Pathogenesis and treatment of varicoceles. BMJ 2004; 328: 967-968
Tekgul S, Dogan HS, Hoebeker P, et al. Guidelines on Pediatric Urology. London: Eur Soc Paediatr Urol, 2016
Vijayaraghavan SB. Sonographic differential diagnosis of acute scrotum. J of Ultrasound in Medicine 2006: 25: 563-574
Wright LA, Gerscovich EO, Corwin MT, et al. Tension hydro- cele: additional cause of ischemia of the testis. J of Ultrasound in Medicine 2012; 31: 2041-2043
Wu, S, Liu G, Chen S, et al. Sonographic patterns of testicular torsion. J of Diagnostic Medical Sonography 2011; 27: 273-278