Definição
A enterocolite necrosante (ECN) é uma situação clínica gastrintestinal de repercussão sistémica e gravidade progressiva, afectando sobretudo o recém-nascido pré-termo; é caracterizada fundamentalmente por vários graus de necrose da mucosa ou transmural do intestino, em áreas de extensão variável, no íleo terminal (mais frequentemente), cólon ascendente e porção proximal do cólon transverso.
De etiopatogénese não totalmente esclarecida, admite-se a comparticipação de múltiplos factores culminando num processo agudo de isquémia-reperfusão associado a uma resposta inflamatória amplificada em concomitância com processo infeccioso decorrente da invasão de microrganismos.
Aspectos epidemiológicos e importância do problema
Esta afecção – que constitui a emergência cirúrgica mais frequente no recém-nascido – atinge com maior frequência os recém-nascidos pré-termo (com menos de 37 semanas completas contadas a partir do 1º dia da última menstruação), principalmente os de muito baixo peso (inferior a 1.500 gramas). A ECN surge em 5 a 15% dos recém-nascidos pré-termo hospitalizados em UCIN; a incidência é máxima entre a primeira e segunda semana. Nos Estados Unidos, a incidência oscila entre 1 a 3 casos por 1000 nados-vivos, com maior número de casos entre a primeira e segunda semana de vida, sem predomínio de sexo; considerando a globalidade dos casos, cerca de 7% corresponde a RN de termo.
Em Portugal, no âmbito da Secção de Neonatologia da SPP foi realizado um estudo multicêntrico pelo Grupo de Registo Nacional dos Recém-Nascidos de Muito Baixo Peso (RNMBP ou de peso < 1.500 g) no quinquénio 1996-2000, englobando unidades neonatais de 35 hospitais nacionais. Estudada a série de 4355 RN com tais características ponderais, o diagnóstico de ECN ocorreu em 437 (~10%). A letalidade nos RN com ECN perfurada foi de 64,3% quando não submetidos a cirurgia, e de 29,5% quando operados.
Na UCIN (médico-cirúrgica) do Hospital de Dona Estefânia (Lisboa) foram assistidos 114 RN com o diagnóstico de ECN no período de 22 anos (1990 a 2011). Globalmente a taxa de mortalidade foi de 27,2%, a que corresponde letalidade de 14,9% e taxa de sequelas (estenose intestinal e síndroma de intestino curto) de 45,3%. De referir que nesta série de RN com ECN, 41% dos doentes tinham peso inferior a 1.000 g.
Etiopatogénese
Apesar de ainda existirem muitos enigmas quanto à patogénese da ECN, admite-se que na generalidade dos casos o factor major envolvido é a prematuridade, dado que a incidência e gravidade desta patologia é inversamente proporcional à idade gestacional.
No RN pré-termo (RNPT), e na base da imaturidade intestinal, estão determinadas características tais como maior permeabilidade da barreira resultante da débil união entre enterócitos, menor secreção de muco e de IgA, motilidade diminuída e maior vulnerabilidade a determinadas agressões. Com efeito, verificou-se que determinadas agressões que alteram o tono do leito microvascular intestinal (hipóxico-isquémicas, infecciosas, relacionadas com a introdução de alimentação entérica, etc.) desencadeiam uma série de reacções inflamatórias em cascata, desproporcionais e excessivas, associadas a invasão da respectiva mucosa por agentes microbianos com consequente proliferação. Este processo, que culmina em necrose de coagulação das áreas afectadas, tipifica a patogénese “clássica” (90% dos casos), afectando essencialmente a ECN nos RN de muito baixo peso (RNMBP).
Por outro lado, determinados estudos sobre microbiota intestinal permitiram identificar, em cerca de 10% dos casos, outro grupo de ECN, afectando RN de termo (RNT) que tipicamente evidenciam quadro clínico iniciado em fase mais precoce e relacionado predominantemente com patologia ao nível do cólon. Os factores de risco associados a este grupo de RNT incluem designadamentre: restrição do crescimento fetal, asfixia perinatal, cardiopatia congénita, policitémia, gastrosquise, exsanguinotransfusão, transfusão de hemoderivados, sépsis, cateterismo umbilical, ruptura prematura de membranas, corioamnionite e diabetes gestacional.
Seguidamente são abordados os principais factores etiopatogénicos, interligados, como que num círculo vicioso. A separação por alíneas foi feita por razões didácticas
Circulação intestinal e isquémia
Admite-se que a hipóxia-isquémia intrauterina promove a redistribuição do débito cardíaco em favor do coração e do sistema nervoso central, privando o intestino imaturo de oxigenação adequada. Efectivamente, pela avaliação do fluxo sanguíneo através do método doppler, demonstrou-se redução do fluxo sanguíneo na artéria mesentérica superior e no tronco celíaco nas situações de restrição de crescimento intrauterino. Esta alteração mantém-se após o nascimento durante a primeira semana de vida, o que sugere, segundo alguns investigadores, que a maior resistência vascular mesentérica já venha programada desde a vida intrauterina. Noutros estudos comprovou-se que a reticulocitose no recém-nascido pré-termo com restrição de crescimento intrauterino constitui um marcador de maior risco para o desenvolvimento de ECN.
Relativamente às características da circulação neonatal cabe referir que existe um equilíbrio muito lábil entre vasodilatação e vasoconstrição, fenómenos mediados respectivamente pelo óxido nítrico (NO) e pela endotelina-1. O estado neonatal basal sob o ponto de vista fisiológico é caracterizado pelo predomínio do NO, gerando-se baixa resistência vascular sistémica. Os estados patológicos causam disfunção endotelial, o que conduz a activação da endotelina-1 e a vasoconstrição, isquémia intestinal e lesão celular. Este mecanismo é compatível com os achados histológicos de necrose de coagulação, típicos da ECN.
Por outro lado, embora o NO desempenhe papel importante na homeostase do tracto gastrintestinal, em situações associadas a inflamação é produzido em elevadas concentrações, o que tem efeito citotóxico directo nos enterócitos.
Substrato
O crescimento e o desenvolvimento do tubo digestivo, assim como a sua capacidade em manter as funções de digestão e absorção, dependem do suprimento adequado em vários nutrientes. A arginina, aminoácido que pode ser sintetizado pelo enterócito, constitui a principal fonte de azoto para a produção local de óxido nítrico. Por sua vez, o óxido nítrico funcionando como mediador-indutor do relaxamento da musculatura lisa vascular, contribui para regular o tono basal arteriolar e, por consequência, o débito sanguíneo ao nível da mucosa intestinal.
A propósito, é importante mencionar estudos experimentais provocando hipóxia-isquémia, ou administrando toxinas ou factor de activação plaquetário; os mesmos demonstraram que a inibição da síntese de óxido nítrico se associou a maior intensidade da lesão tecidual. Por outro lado, comprovou-se que o suprimento exógeno de óxido nítrico contribuiu para atenuar tal efeito. Noutros estudos experimentais demonstrou-se também que a suplementação em arginina (por via oral ou por via endovenosa contínua) atenuava a lesão intestinal na sequência de eventos hipóxico-isquémicos seguidos de reoxigenação.
Por outro lado, há que atender ao facto de a imaturidade intestinal não permitir a absorção e digestão completas dos hidratos de carbono e gorduras do leite. Como consequência, os compostos não digeridos servem como substrato para a proliferação de bactérias entéricas, do que resulta acumulação de hidrogénio, ácidos orgânicos, caseína não digerida e ácidos gordos de cadeia longa no lume intestinal. Admite-se que exposição do epitélio intestinal a estas substâncias origine um processo de inflamação intestinal, conduzindo a lesão.
Imaturidade intestinal e alimentação entérica
Embora se admita classicamente que a colonização do tracto intestinal por germes microbianos constitua um pré-requisito para o desenvolvimento de ECN, a doença pode surgir em crianças sem terem sido alimentadas previamente (cerca de 5-7% dos casos).
O Quadro 1 resume algumas das características que permitem definir a imaturidade gastrintestinal (cuja expressão máxima se verifica no recém-nascido pré-termo) e as consequências que daí resultam.
QUADRO 1 – Imaturidade intestinal e consequências.
| Défice de secreção gástrica e hipocloridria Colonização bacteriana aberrante do tracto gastrintestinal superior; digestão proteica incompleta |
| Défice de enzimas proteolíticas Défice de destruição das toxinas bacterianas; digestão proteica incompleta |
| Motilidade intestinal diminuída Estase e hipercrescimento bacteriano |
| Défice de secreção de IgA Alteração do mecanismo de defesa contra antigénios bacterianos |
| Redução do número de linfócitos T intestinais Alteração do mecanismo de preservação da integridade do epitélio intestinal por incapacidade de destruição das células epiteliais infectadas |
| Hiperpermeabilidade da mucosa intestinal a proteínas, hidratos de carbono e bactérias Acesso facilitado de bactérias e toxinas aos tecidos intestinais |
Cabe referir que a alimentação com leite materno fresco constitui uma circunstância susceptível de proteger contra lesões do intestino, tendo em conta a multiplicidade de factores imunoprotectores que o referido leite veicula. De facto, diversos estudos têm demonstrado menor incidência de ECN em crianças alimentadas com leite materno. Estudos em recém-nascidos pré-termo também levaram à conclusão de que a modificação do leite não materno através, designadamente, da acidificação (pH entre 2,5 e 5,5) diminui a taxa de colonização bacteriana gástrica.
Microbiota e toxinas bacterianas
Relativamente a estes factores, importa realçar alguns aspectos:
- As bactérias intestinais comensais, de acção benéfica, que integram o microbioma intestinal, (Bifidobacterium, Lactobacillus e Bacteroides) mantêm a homeostase intestinal, a função imune e facilitam a digestão, ao mesmo tempo que protegem o intestino contra a inflamação e diversas agressões, já referidas.
Nesta perspectiva, importa citar algumas situações que influenciam, atrasam ou comprometem tal colonização benéfica (RN pré-termo, alimentação com fórmula, antibioticoterapia empírica, parto por cesariana, entre outras) e referir que a par da taxa diminuída de colonização dos comensais, surge aumento da prevalência de Clostridium perfringens, Proteobacteria, Firmicutes e Enterobacter. Portanto, situação de disbiose.
Ora, em diversos estudos, verificou-se que tal alteração no microbioma precede o surgimento da ECN.
Quanto ao papel das enterobactérias Gram-negativas (E. Coli, Klebsiella, Proteus, etc.) admite-se que actuem:
- Através de endotoxina com característica de fraca citotoxicidade directa, mas causando lesão tecidual difusa activando a cascata inflamatória; e, indirectamente,
- Através da disfunção dum receptor para os Gram-negativos; tal receptor, reconhecendo o padrão molecular associado a tais agentes microbianos, como que “barra, faz barragem ou “trava”, como nas “portagens”, a invasão daqueles Gram-negativos. Daí a designação de “Toll-Like Receptor 4” ou TLR-4. Por consequência, a disfunção deste contribui para resposta inflamatória excessiva e lesiva para o intestino.* Pelo contrário, a microbiota comensal (Bifidobacterium, Lactobacillus e Bacteroides) tem efeito contrário, diminuindo a inflamação.
*O Toll-like receptor 4 (Receptor TLR-4) é uma proteína codificada pelo gene TLR4. Reconhecendo determinados compostos como por exemplo o lipopolissacárido (LPS), um componente presente em muitas bactérias Gram-negativas, é responsável pela activação do sistema imune inato. |
- Os germes bacterianos mais frequentemente implicados como causa específica de ECN são algumas espécies de Clostridia (difficile, perfringens), as quais infectam com especial preferência o tecido isquémico, sendo relevante o papel de toxinas que produzem; de referir, no entanto, que as Clostridia fazem parte da microbiota do cólon do recém-nascido. A este propósito importa salientar que os RN pré-termo evidenciam uma resposta inflamatória excessiva aos micróbios luminais o que contribui para fragilizar a barreira protectora do intestino.
- Staphylococcus coagulase negativo e Staphylococcus aureus produzindo toxinas citolíticas (delta toxinas provocando lesão celular intestinal) têm sido considerados nalguns estudos importantes agentes patogénicos.
- Os germes microbianos isolados a partir do líquido peritoneal (bactérias e vírus, fungos) dos doentes com ECN são representativos, por um lado, da microbiota do cólon e, por outro, da transferência dos mesmos a partir do intestino lesado.
- Relativamente aos agentes víricos, cabe referir Coxsackie B2, Coronavirus e Rotavirus, descritos como desencadeadores de quadro de ECN.
Em suma, não se poderá responsabilizar determinado germe especificamente pelo desenvolvimento de ECN, embora se tenha demonstrado papel importante dalguns deles em circunstâncias de surtos epidémicos.
Mediadores inflamatórios
Uma referência sucinta ao papel dos mediadores inflamatórios locais cuja produção pode ser desencadeada pela colonização aberrante e pela inadequada neutralização de toxinas atrás referidas. Tal atipia do padrão de colonização, associada a imaturidade do epitélio intestinal, origina uma resposta inflamatória bacteriana com produção excessiva de citocinas pró-inflamatórias, sendo que parte desta resposta se relaciona com o sistema imune inato.
A resposta é iniciada com produtos moleculares derivados da parede celular bacteriana actuando sobre receptores presentes no epitélio intestinal iniciando-se a activação da cascata inflamatória na qual tomam parte mediadores inflamatórios como o PAF, TNF-alfa, interleucinas 1, 6, 8, 12, 18, NO, LPS, e radicais livres de oxigénio. Em doentes com ECN os níveis de citocinas estão elevados, correlacionando-se com a gravidade da doença.
O factor de activação das plaquetas (PAF ou platelet-activating factor, regulado pela enzima com efeito de degradação acetil-hidrolase PAF-AH), um fosfolípido, é produzido por células endoteliais, neutrófilos, macrófagos, próprias plaquetas, como resposta a endotoxinas e hipóxia.
O factor de necrose tumoral-alfa (TNF-alfa ou tumor necrosis factor-alpha) é uma citocina libertada por macrófagos sobre os quais actuam endotoxinas.
Ao nível do intestino, a produção de mediadores inflamatórios activando os neutrófilos, originando vasoconstrição, lesão dos vasos capilares intestinais e hipotensão, promove a libertação de radicais livres com consequente lesão intestinal que pode culminar em necrose. De acordo com diversos estudos, o PAF causa lesão intestinal por via dos radicais livres de oxigénio. (ver atrás – o papel dos Toll-like receptors-4: TLR-4)
Outro importante mediador é o chamado lipopolissacárido (LPS), a endotoxina componente das bactérias Gram-negativas, abundantes no tracto gastrintestinal. O mesmo altera a função da barreira gastrintestinal, promovendo a libertação doutros mediadores inflamatórios como NO, interferão – gama e cicloxigenase, com efeitos tóxicos directos sobre os enterócitos.
A fosfatase alcalina intestinal, enzima produzida pelos enterócitos destoxifica o LPS, tendo sido concluído em estudos diversos que a probabilidade de ECN é maior nos doentes em que a fosfatase alcalina (FA) intestinal é deficiente. Daí a especulação quanto ao eventual papel preventivo e terapêutico da mesma.
Lesão por isquémia-reperfusão e acção dos radicais livres de oxigénio
A isquémia seguida de reperfusão do intestino origina aumento da permeabilidade da membrana das células intestinais (atrás referida) e incremento de produção de radicais livres de oxigénio com consequente lesão da referida membrana através de processo de peroxidação lipídica.
Embora o recém-nascido evidencie capacidade limitada para a produção de radicais livres de oxigénio (através da acção das enzimas xantina-oxidase e NADPH-oxidase dos neutrófilos), a capacidade de destoxificação daqueles (através das enzimas catalase, superóxido – dismutase e glutationa – peroxidase) é ainda mais limitada, o que aumenta a probabilidade de lesão do intestino.
A lesão celular, ocorrendo também ao nível do endotélio vascular, pode resultar ainda em perda da integridade deste, agravando os fenómenos de isquémia nos territórios de circulação mesentérica de tipo terminal, como a observada na região ileal distal e da válvula íleo-cecal, a qual é irrigada pela artéria ileocólica, ramo terminal da artéria cólica direita.
Fármacos e substâncias tóxicas
Os fármacos e substâncias tóxicas mais frequentemente associados ao aparecimento de ECN (xantinas e metilxantinas, vitamina E, indometacina, etc.) comportam, de facto, um risco acrescido pela alteração do lábil equilíbrio hemodinâmico e vasomotor em recém-nascidos evidenciando grau importante de imaturidade, o que favorece o desencadeamento de fenómenos vasoclusivos.
Factor de crescimento epidérmico
Demonstrou-se que os factores de crescimento desempenham importante papel, não só no desenvolvimento do tracto gatrintestinal, como na resposta às agressões.
O chamado factor de crescimento epidérmico (FCE) é um péptido que pertence a uma família que inclui outros péptidos, responsável por um conjunto de respostas biológicas no tubo digestivo dizendo respeito, essencialmente, à regulação da replicação celular, e ao movimento e à sobrevivênvia das células.
Esta família de péptidos tem afinidade com receptores específicos (receptores do FCE) distribuídos em vários territórios do organismo e ao longo do tubo digestivo do feto e RN; mais concretamente, tais receptores localizam-se, respectivamente, no compartimento basolateral das células da epiderme e na membrana apical do epitélio viloso intestinal.
Estudos recentes relacionam tal FCE com a ECN verificando, designadamente, excreção urinária de FCE em RN com quadro de ECN, especulando-se que tal resulta de maior absorção de FCE no intestino lesado. Outros estudos, apontando a associação entre níveis baixos de FCE na saliva e no soro, e o aparecimento de ECN, levantam a hipótese de a administração daquele ter importância na prevenção e tratamento.
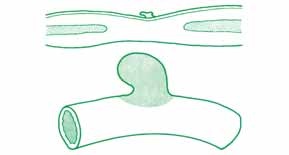
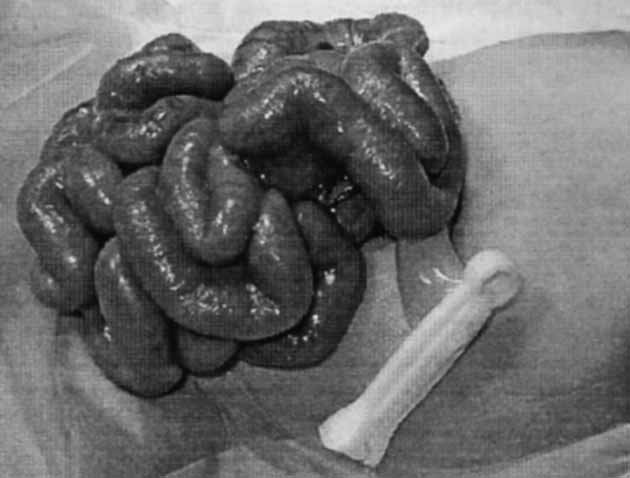
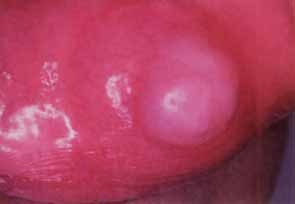
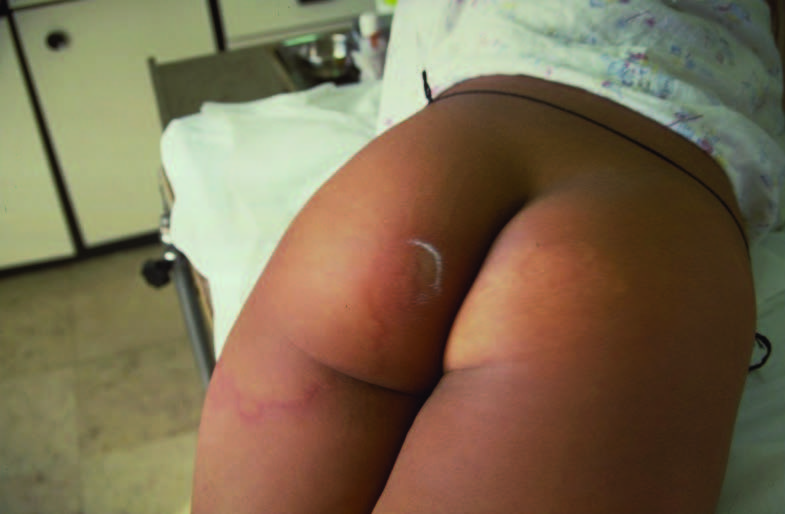
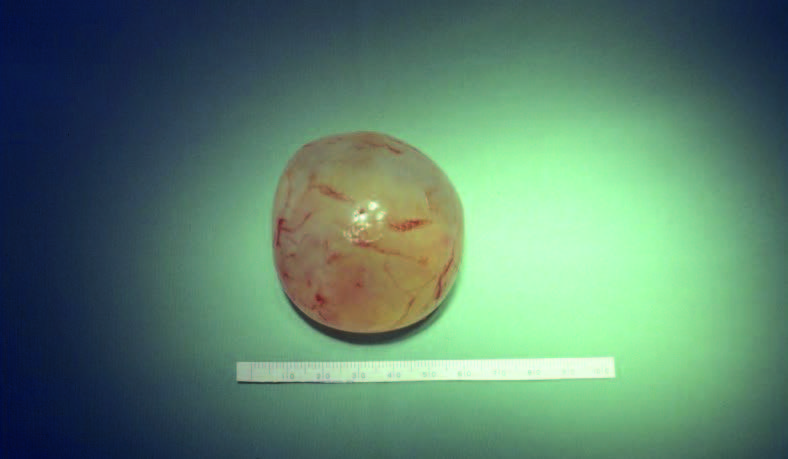
Como consequência anatomopatológica das diversas noxas descritas, o exame macroscópico das ansas revela que as mesmas estão distendidas e com paredes friáveis; a mucosa evidencia áreas hemorrágicas ulceradas e necrosadas, podendo estar cobertas por exsudado seroso. Faz parte do quadro a verificação de gás intramural (de localização subserosa ou submucosa) denominada pneumatose). A Figura 1 (achado intra-operatório) é elucidativa: imagens esféricas simulando “pequenos balões” ao nível da parede intestinal, os quais têm tradução radiológica. (ver adiante)
Pode verificar-se igualmente líquido peritoneal, claro, turvo ou hemorrágico, aspectos que variam em função do grau de inflamação.
O exame histológico da parede intestinal pode evidenciar aspectos variáveis: edema, áreas hemorrágicas e de necrose de coagulação, úlceras, áreas de trombose, e sinais de reparação tecidual. As áreas lesadas estão cobertas por células inflamatórias, fibrina, e epitélio necrótico que, conglomerados em camada, formam uma pseudomembrana. Nalgumas situações pode verificar-se gás no sistema porta. Como característica relevante, refere-se a concomitância de áreas de inflamação, necrose e reparação teciduais, o que testemunha as características evolutivas desta entidade clínica.
Manifestações clínicas e diagnóstico
Como factores predisponentes mais típicos nos RN pré-termo são referidos os seguintes: infecção materna, ruptura de membranas > 24 horas antes do parto, ductus arteriosus permeável e sintomático, asfixia perinatal, acidose, choque, alimentação entérica com fórmula, administração de ranitidina, etc..
Nos RN de termo e/ou de peso superior a 2.000 gramas apontam-se os seguintes factores predisponentes: asfixia perinatal, policitémia/ hiperviscosidade, problema respiratório, hipoglicémia, antecedentes de intervenção cirúrgica abdominal para correcção de defeitos da parede abdominal ou de lesões do tubo digestivo, cardiopatia congénita de baixo débito sanguíneo no território intestinal, etc..
O quadro clínico pode variar entre uma forma benigna, subclínica, com recuperação total sem sequelas, até uma forma grave, com sinais de sépsis, choque, peritonite generalizada, coagulopatia e falência multiorgânica.
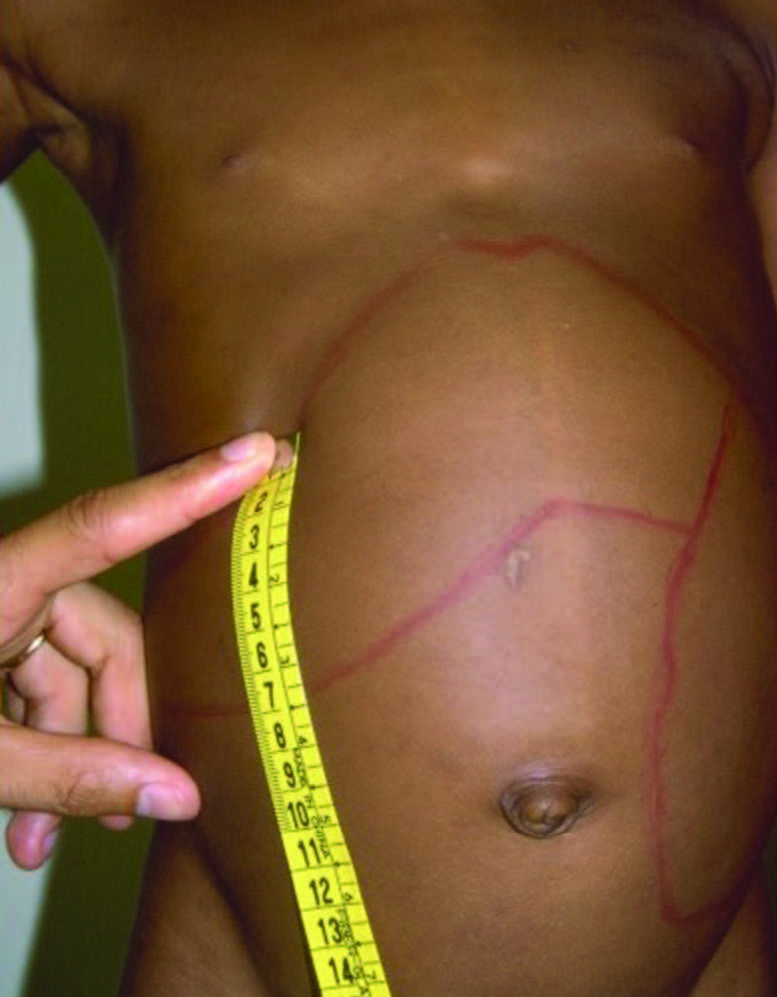
Os sinais clínicos mais característicos incluem: distensão abdominal, dificuldade respiratória, resíduo gástrico, vómito bilioso, diarreia, rectorragia, dificuldade respiratória, labilidade hemodinâmica e térmica, e alterações inflamatórias da parede abdominal com eritema e rede venosa visível, indicativas de peritonite e de necrose intestinal subjacente.
A palpação abdominal pode evidenciar hiperestesia localizada com empastamento subjacente secundário a sofrimento de ansa abdominal ou massa abdominal, relacionável com aglomerado de ansas imóveis, o qual pode indiciar perfuração coberta ou abcesso intraperitoneal. A distensão extrema e a presença de sinais peritoneais generalizados são compatíveis com necrose transmural e perfuração de ansa, peritonite grave e pneumoperitoneu.
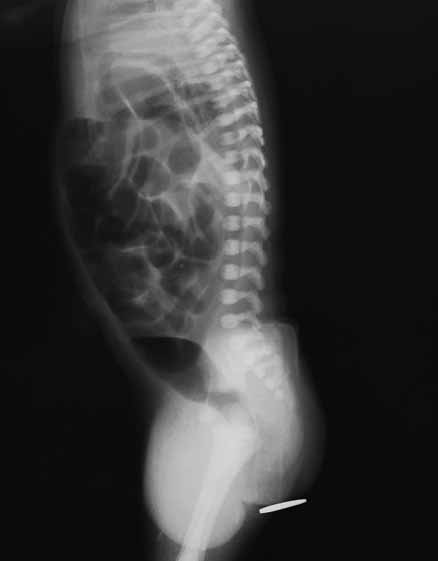
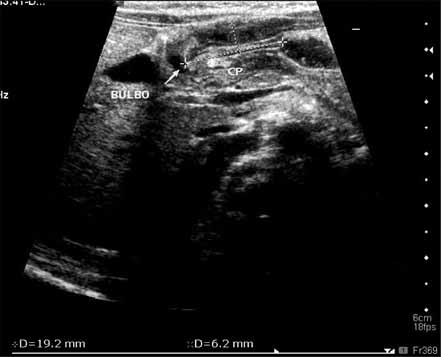
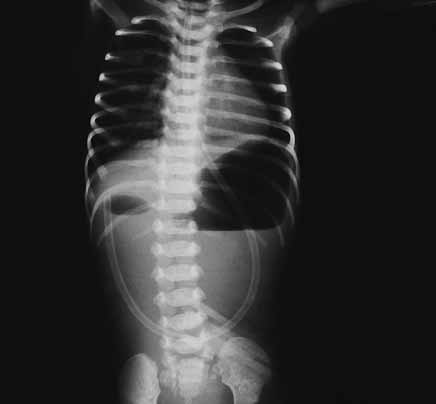
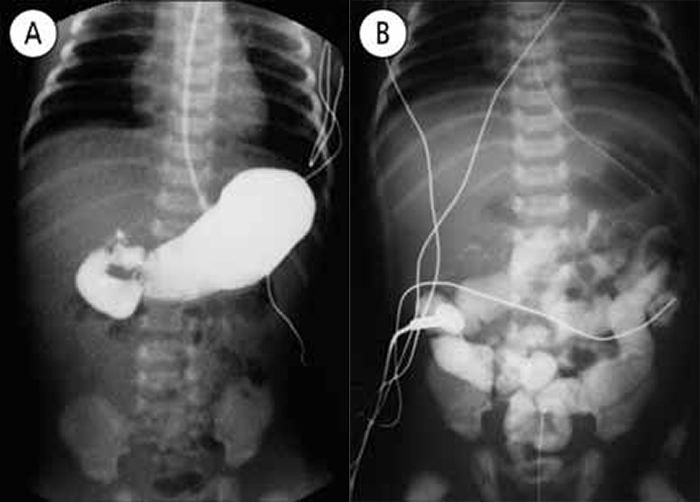
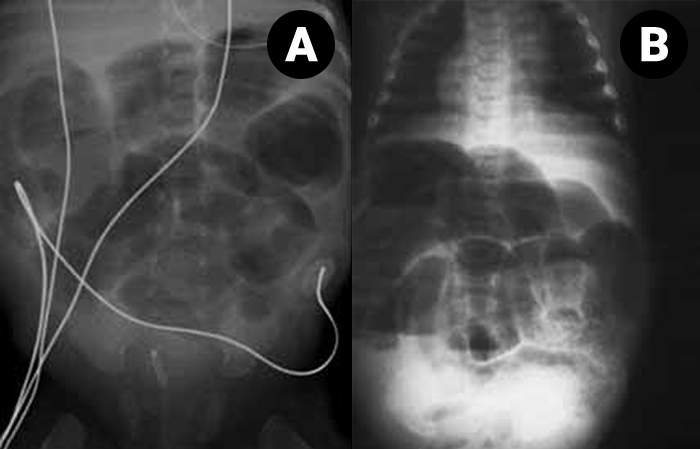
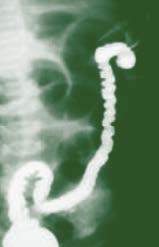
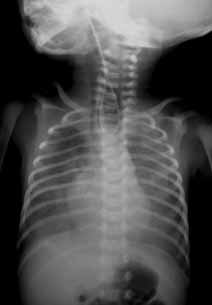
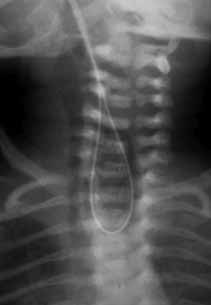
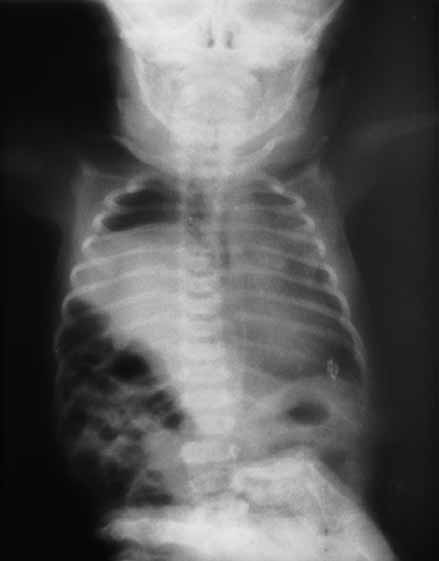
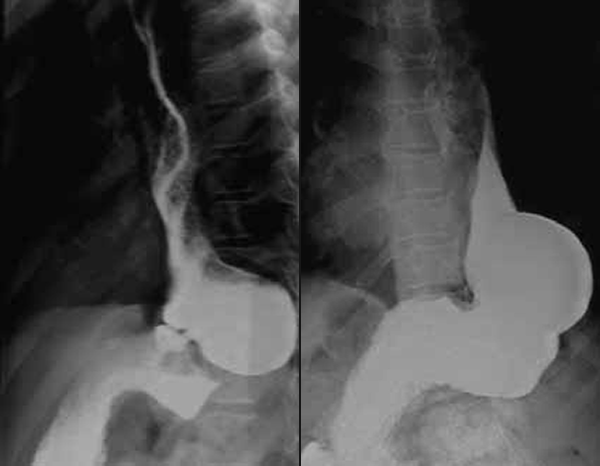
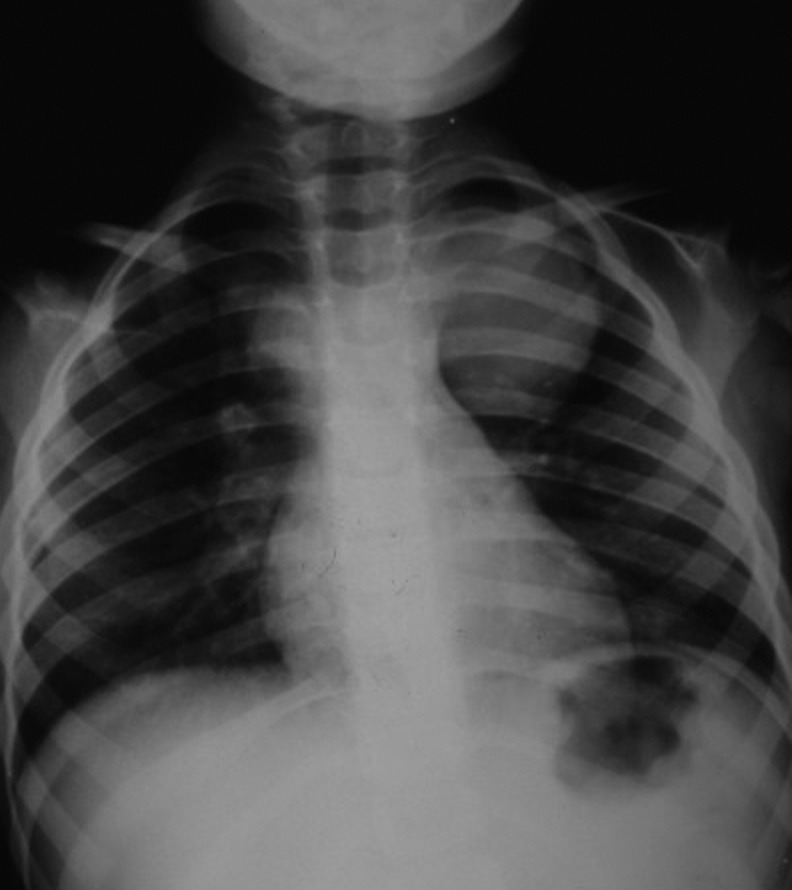
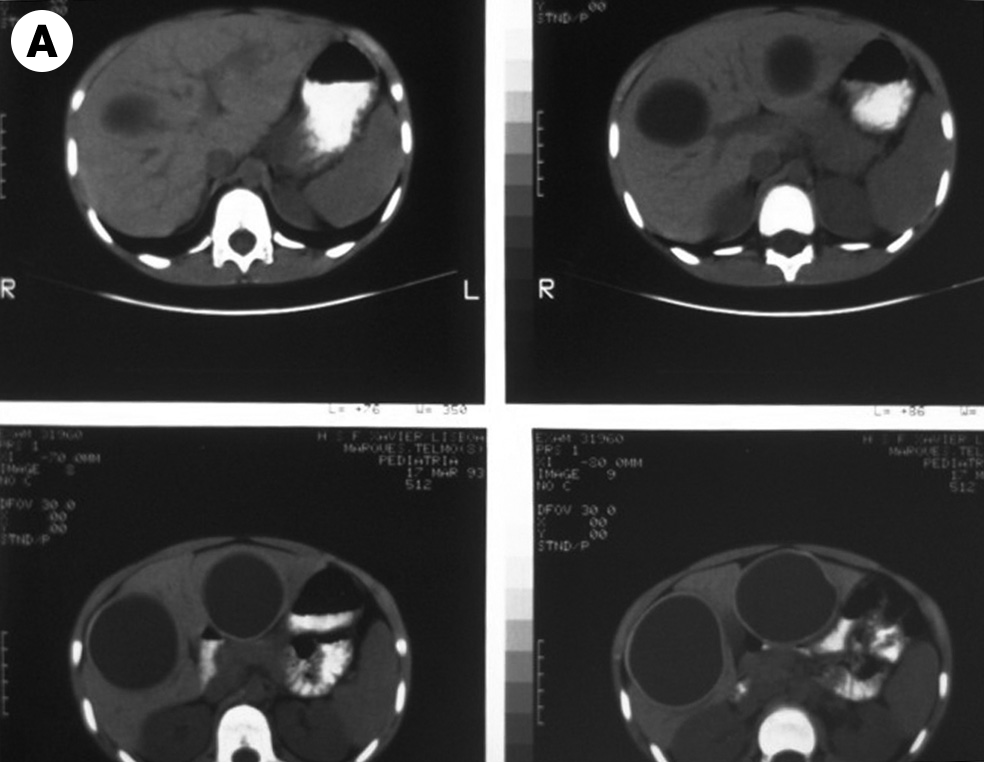
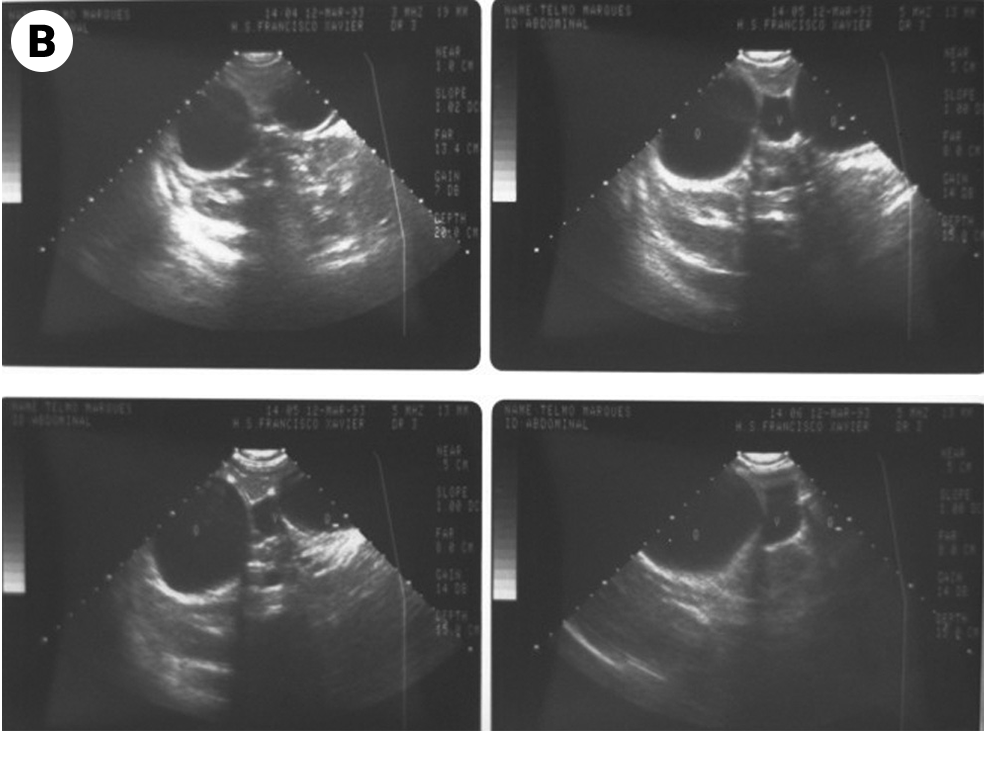
O diagnóstico radiológico de ECN está bem determinado, salientando-se os principais sinais: distensão de ansas; presença de gás intramural (pneumatose intestinal); ascite; pneumoperitoneu; presença de gás na circulação porta; ansa intestinal edematosa e fixa; diminuição de gás intrabdominal com presença de ansas assimétricas e distensão cólica. (Figuras 2 a 4)
Torna-se óbvio concluir que a vigilância imagiológica deve ser seriada para comparação evolutiva dos padrões anómalos identificados.
Como manifestações laboratoriais frequentemente associadas enumeram-se as mais importantes: neutropénia ou neutrofilia com aparecimento de formas imaturas no sangue periférico, trombocitopénia, perfil de coagulação anómalo, hiponatrémia de aparecimento súbito, acidose metabólica, hipoproteinémia, hiperglicémia, etc..
Sob o ponto de vista da evolução clínica e gravidade são descritos diversos estádios definidos por Bell (Critérios evolutivos de Bell) cuja identificação, valorizando de modo cumulativo sinais sistémicos, intestinais e imagiológicos, tem implicações práticas importantes quanto às decisões terapêuticas e ao prognóstico. (Quadro 2)
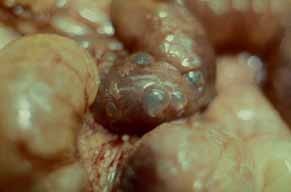
FIGURA 1. Aspecto macroscópico de pneumatose (gás intramural); distensão bolhosa. (NIHDE)
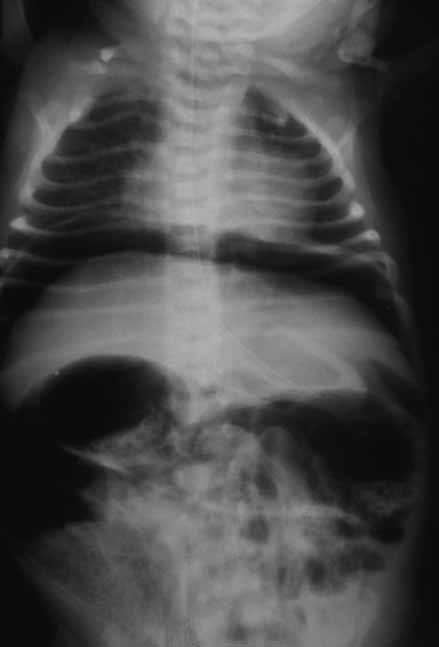
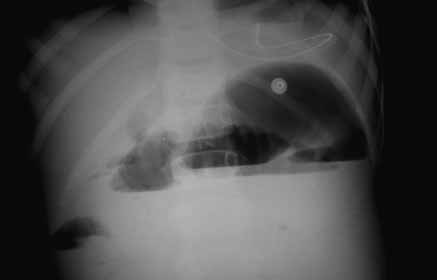
FIGURA 2. ECN – Imagem radiológica de pneumoperitoneu.
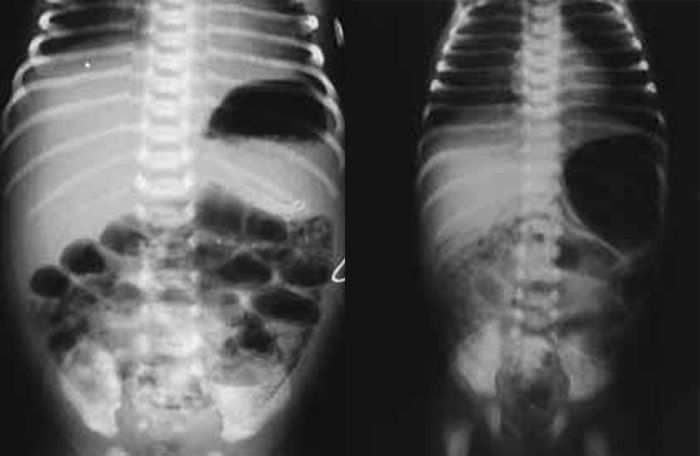
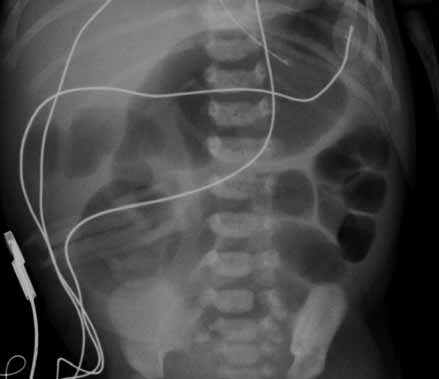
FIGURA 3. ECN – Imagem radiológica abdominal simples evidenciando distensão abdominal, edema e espessamento da parede das ansas e sinais de pneumatose (gás intramural); presença de gás na área hepática.
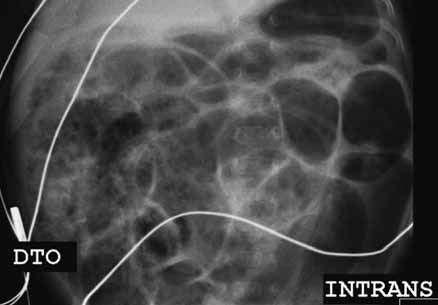
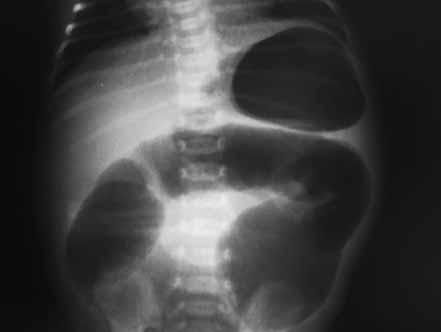
FUGURA 4. ECN – Imagem de radiografia abdominal simples com sinais de pneumatose e panecrose.
QUADRO 2 – Estádios Evolutivos de Bell na ECN.
I A – Suspeita de ECN |
I B – Suspeita de ECN |
II A – ECN definida (forma ligeira) |
II B – ECN definida (forma moderada) |
III A – ECN avançada (forma grave) |
III B – ECN avançada (forma grave com perfuração intestinal) |
Para avaliação do grau de oxigenação ao nível dos órgãos (designadamente SNC, intestino, e outros), com valor prognóstico, presentemente (2020), alguns centros dispõem de tecnologia aplicando espectroscopia próxima dos infra-vermelhos.
Prevenção
Ao delinear estratégias de prevenção torna-se fundamental entrar em conta com os mecanismos potencialmente envolvidos na etiopatogénese.
Ao longo do tempo têm sido preconizadas diversas estratégias, algumas das quais têm evoluído em função dos resultados da investigação científica, designadamente de meta-análises de uma multiplicidade de estudos.
Seguidamente são descritas diversas medidas, as quais foram estratificadas em função de critérios de segurança e eficácia, com base em estudos da Cochrane Library.
1 – Leite materno
O leite humano contém múltiplos factores tais como imunoglobulinas, interleucina-10, FCE, acetil-hidrolase, entre outros; por outro lado, o factor de activação plaquetária (PAF) que comparticipa a etiopatogénese da ECN evidencia concentrações elevadas em casos de ECN, enquanto os níveis da enzima que promove a sua hidrólise (acetil-hidrolase) estão diminuídos.
Ora, o leite humano contém níveis elevados de FCE e de acetil-hidrolase, factores que são protectores em relação à ECN. Daí a incidência cerca de 6 a 10 vezes menor de ECN em RN pré-termo alimentados com leite materno, em comparação com a verificada nos alimentados com fórmula, o que tem sido provado em estudos de meta-análise.
Trata-se duma medida segura, de eficácia comprovada. Deve ser iniciada nos primeiros 2-5 dias (a pausa alimentar > 5-7 dias, inicialmente recomendada é actualmente desaconselhada), recomendando-se nos primeiros dias alimentação entérica trófica com leite materno, não nutricional, com incrementos diários modestos e prudentes ~15 mL/kg/dia, em função da avaliação clínica caso a caso. (ver Parte sobre Neonatologia)
2 – Corticoterapia pré-natal
Uma vez que o nascimento antes do termo da gravidez constitui o factor de risco mais relevante de ECN, a possibilidade de indução medicamentosa da maturidade intestinal com a utilização de corticosteróide pré-natal tem sido estudada. A este propósito cabe referir que os resultados de estudos aleatórios multicêntricos não têm sido concordantes: nalguns demonstrou-se diminuição de incidência de ECN, enquanto noutros, precisamente o contrário.
Apesar destes achados aparentemente contraditórios, a utilização de corticóides pré-natais (betametasona) está hoje consagrada como uma importante medida para a redução da mortalidade e morbilidade relacionáveis com a imaturidade pulmonar e com a prematuridade em geral.
Esta medida é considerada de eficácia comprovada, embora de segurança questionável.
3 – Encerramento precoce do ductus arteriosus
A presença de canal arterial patente promove um desvio do volume sanguíneo para as artérias pulmonares na fase diastólica, o que tem como consequência a diminuição da perfusão do territórios esplâncnico, aumentando a probabilidade de ECN. Este dado fisiopatológico tem confirmação na prática clínica na sequência de estudos controlados e aleatórios em recém-nascidos pré-termo de peso inferior a 1.000 gramas submetidos a laqueação cirúrgica precoce do canal arterial.
A partir do início dos anos 80, a indometacina (inibidor das prostaglandinas) passou a ser usada profilacticamente, com eficácia demonstrada, para o encerramento do canal arterial e prevenção da hemorragia intracraniana. No entanto, estudos ulteriores identificaram efeitos colaterias, tais como diminuição do fluxo sanguíneo esplâncnico, aumento da incidência de ECN e perfuração intestinal, comprometendo a recomendação universal para o seu uso.
Noutros estudos demonstrou-se diminuição do fluxo sanguíneo esplâncnico menos marcada empregando outro fármaco, também inibidor das prostaglandinas – o ibuprofeno. Recentemente, dados da Cochrane Library provaram que o uso de indometacina não está associado a aumento de risco de ECN.
Esta medida é considerada de eficácia comprovada, embora de segurança questionável.
4 – Antibioticoterapia por via enteral
A análise de estudos da Cochrane Library empregando antibióticos por via enteral (aminoglicosídeos) sugere, de facto, que tal procedimento contribui para reduzir tanto a incidência, como a mortalidade por ECN. No entanto, face ao risco acrescido de selecção de estirpes com tal estratégia, tal procedimento não deve ser posto em prática.
Esta medida é considerada de eficácia comprovada, embora de segurança questionável.
5 – Probióticos
Na sequência do que foi referido no capítulo respeitante a esta área, importa acentuar que, em modelos experimentais e em estudos meta-analíticos na espécie humana, se comprovou a eficácia na prevenção no RN pré-termo, com redução da incidência e da mortalidade, recomendando-se o uso de duas ou mais espécies, incluindo designadamente Lactobacillus acidophilus e Bifidusbacterium spp. Contudo, os investigadores alertaram para o risco de sépsis em RN pré-termo com peso < 750 gramas.
Tendo em consideração que os estudos analisados adoptaram metodologias diversas, considera-se que esta medida é considerada de eficaz, embora de segurança questionável.
6 – Suplemento de arginina
Com base no achado anátomo-patológico de necrose de coagulação, resultante de eventos isquémicos locais ou sistémicos, o papel do óxido nítrico tem adquirido importância especial. Com efeito, o óxido nítrico é produzido durante a conversão enzimática da L-arginina em L-citrulina sob a acção da sintetase de NO. Embora os investigadores considerem o suplemento exógeno de arginina uma arma promissora na prevenção da ECN, esta medida é considerada de eficácia comprovada, embora de segurança questionável.
7 – Novos fármacos (anticitocinas e factores de crescimento)
Com estes fármacos, em fase de investigação, foi demonstrada eficácia em modelos animais, mas não na espécie humana.
8 – Pré-bióticos (derivados do leite humano e de plantas), glutamina, ácidos gordos ómega-3, receptores agonistas funcionando como barreira a compostos microbianos
Com estes compostos, também em fase de investigação, não foi demonstrada eficácia, razão pela qual são desaconselhados.
9 – Imunoglobulinas e bloqueadores H2 por via oral
Sabendo-se que no recém-nascido pré-termo são baixos os níveis séricos de imunoglobulinas, nomeadamente IgA secretória, diversos estudos aleatórios avaliaram o papel da utilização profiláctica de preparados de imunoglobulinas e de bloqueadores H2 por via oral na prevenção da ECN.
Embora alguns autores tivessem comprovado redução significativa da doença nos grupos tratados com os referidos compostos, de acordo com a meta-análise da Cochrane Library concluiu-se que são desaconselhados.
Tratamento
1 – Medidas gerais
Perante a suspeita de ECN há que pôr em execução um conjunto de medidas gerais prioritárias de carácter conservador, no pressuposto de que a avaliação, em centro especializado e em unidade de cuidados intensivos, deverá ser feita por equipa multidisciplinar: interrupção imediata de alimentação por via entérica, descompressão gástrica com introdução de sonda naso ou orogástrica, manutenção, após correcção, dos equilíbrios hidroelectrolítico, ácido-base, hemodinâmico, início de nutrição parentérica, início de antibioticoterapia de largo espectro para cobertura de germes gram-positivos, gram-negativos e anaeróbios (esquema empírico a modificar em função do contexto clínico-microbiológico: ampicilina + aminoglicosídeo ou cefalosporina de terceira geração + clindamicina ou metronidazol). No âmbito da avaliação de parâmetros hematológicos, haverá que manter hematócrito em torno de 40-45% e número de plaquetas acima de 40.000/mmc.
Nos casos com boa resposta às medidas gerais acima discriminadas, isto é, com diminuição da distensão abdominal, desaparecimento das imagens radiológicas de pneumatose, desaparecimento do resíduo gástrico e da perda de sangue nas fezes, mantém-se pausa alimentar total até ao 12º ou 14º dia de evolução, e reintroduzindo-se de modo muito cauteloso e progressivo, por fases, o suprimento entérico utilizando leite materno ou fórmula de aminoácidos hiposmolar.
Estas medidas, dum modo geral, aplicam-se aos estádios, de I A a II B (classificação de estádios evolutivos de Bell atrás descrita).
2 – Medidas específicas
Pelo contrário, nos casos em que se verifica progressão rápida do quadro clínico de ECN e agravamento global (correspondendo, em geral aos estádios III A e III B), para além de medidas gerais (mais agressivas, incluindo a administração de inotrópicos e a assistência ventilatória), devem ser ponderados dois procedimentos invasivos: paracentese abdominal para drenagem peritoneal simples e/ou laparotomia.
De referir que a decisão da necessidade e do momento adequado da laparotomia deve ser individualizada com base na análise evolutiva dos achados clínicos e imagiológicos. Uma vez que os doentes em causa evidenciam, na maior parte das vezes, estado crítico, a decisão deve ser tomada de preferência, por equipa multidisciplinar: cirurgião, anestesista e pediatra-neonatologista.
Reportando-nos aos estádios de Bell, a detecção de sinais de ascite (estádio II A), implicará, em princípio, drenagem peritoneal, enquanto a detecção de sinais de pneumoperitoneu – indicativo de perfuração de ansa – (estádio III B) implicará laparotomia exploradora com eventual ressecção do segmento afectado, seguida de anastomose primária ou enterostomias.
Nalguns centros cirúrgicos é realizada já laparotomia em presença do estádio III A (ascite sem evidência de pneumoperitoneu) sendo que a tendência actual, no estádio III A, segundo dados da literatura, seja reservar a drenagem peritoneal simples para os casos de idades gestacionais muito baixas e menor peso.
Para além do pneumoperitoneu, outros sinais mais frequentemente associados a perfuração, estabelecendo a indicação de laparotomia são: massa abdominal (indicativa de perfuração coberta ou de abcesso intraperitoneal), alterações inflamatórias da parede abdominal (indicativas de peritonite e de necrose intestinal subjacente), ansa intestinal em posição fixa nas radiografias simples seriadas e presença de ar no sistema porta.
As alterações laboratoriais indicativas de processo clínico em progressão que poderão estabelecer indicação de laparotomia são: alterações da coagulação, trombocitopénia, hiponatrémia e acidose metabólica persistente.
Igualmente, a detecção de germes na coloração pelo Gram no material obtido por paracentese abdominal previamente realizada, poderá constituir indicação para laparotomia.
Uma vez concretizada a ressecção intestinal, deve restabelecer-se, logo que possível, o trânsito intestinal, nomeadamente se em presença de estabilidade hemodinâmica, e na ausência de peritonite ou de ressecção jejunal muito proximal. Nalguns casos há que proceder a duas ou mais enterostomias descompressivas, utilizando os segmentos intestinais viáveis e funcionantes para o restabelecimento ulterior do trânsito intestinal.
No período pós-operatório, o doente deve ser submetido a programa de nutrição parentérica total, pelo que se torna necessário colocar uma via central de longa duração (cateter do tipo Hickman-Broviac).
Prognóstico
As complicações letais da ECN prendem-se com a progressão do processo patológico desencadeante, que pode culminar com o desenvolvimento da chamada síndroma de reacção inflamatória sistémica (SRIS) num contexto de sépsis e acidose metabólica irreversível.
Em cerca de 20 a 25% dos casos poderão desenvolver-se quadros de estenose (fibrose estenosante pós-inflamatória), mais frequente no território ileal distal e cólico; em tais circunstâncias há indicação para ressecção.
A síndroma de intestino curto constitui outra complicação não imediata surgindo como consequência de ressecções intestinais muito alargadas por necrose intestinal extensa.
A sépsis de cateter central, nomeadamente a sépsis por fungos, tem sido apontada como uma complicação relevante pela mortalidade significativa que comporta.
Em suma, os progressos da terapia intensiva e das técnicas operatórias permitem actualmente obter nos casos de ECN uma sobrevivência global > 85%.
Agradecimentos
Os autores e editor agradecem aos Drs. Micaela Serelha, Daniel Virella e Sérgio Pinto a cedência de dados estatísticos e imagiológicos referentes à UCIN-HDE.
BIBLIOGRAFIA
Berman L, Moss RL. Necrotizing enterocolitis: an update. Semin Fetal & Neonatal Med 2011;16:145-150
Blakely ML, Tyson JE, Lally KP, et al. Laparotomy versus peritoneal drainage for necrotizing enterocolitis or isolated intestinal perforation in extremely low birth infants: outcomes through 18 months adjusted age. Pediatrics 2006;117:e680-e687
Burrin DG, Stoll B. Key nutrients and growth factors for neonatal gastrointestinal tract. Clin Perinatol 2002;29:65-96
Coran A. Pediatric Surgery. Philadelphia: Elsevier, 2013
Cuna A, George L, Sampath V. Genetic predisposition to necrotizing enterocolitis in premature infants: current knowledge, challenges, and future directions. Semin Fetal Neonatal Med 2018;23:387-393
Deshpande G, Rao S, Patole S, Bulsara M. Updated meta-analysis of probiotics for preventing necrotizing enterocolitis in preterm neonates. Pediatrics 2010;125:921-930
Frost BL, Caplan MS. Can fish oil reduce the incidence of necrotizing enterocolitis by altering the inflammatory response? Clin Perinatol 2019;46:65-72
Garcia JJ, Cruz O, Mintegi S, Moreno JM (eds). M Cruz Manual de Pediatria. Madrid: Ergon, 2020
Garofalo NA, Caplan MS. Oropharyngeal mother’s milk: state of the science and influence on necrotizing enterocolitis. Clin Perinatol 2019;46:77-89
Goldstein GP, Sylvester KG. Biomarker discovery and utility in necrotizing enterocolitis. Clin Perinatol 2019;46:1-17. https://doi.org/10.1016/j.clp.2018.10.001
Guthmann F, Kluthe C, Buhrer C. Probiotics for prevention of necrotizing enterocolitis. Un updated meta-analysis. Clin Paediatr 2010;222:284-290
Isani MA, Delaplain PT, Grishin A, Ford HR. Evolving understanding of neonatal necrotizing enterocolitis. Curr Opin Pediatr 2018;30:417-423
Kane AF, Bhatia AD, Denning PW, et al. Routine supplementation of Lactobacillus rhamnosus and risk of necrotizing enterocolitis in very low birth weight infants. J Pediatr 2018;195:73-79
Kim CS, Claud EC. Necrotizing enterocolitis pathophysiology: how microbiome data alter our understanding. Clin Perinatol 2019;46:29-38
Kim JH. Role of abdominal US in diagnosis of NEC. Clin Perinatol 2019;46:19-128
Kliegman RM, Willoughby RE. Prevention of necrotizing enterocolitis with probiotics. Pediatrics 2005;115:171-172
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Lin PW, Stoll BJ. Necrotizing enterocolitis. Lancet 2006;368:1271-1283
Long SS, Prober CG, Fischer M (eds). Principles and Practice of Pediatric Infectious Diseases. Philadelphia: Elsevier, 2018
Mangus RS, Subbarao GC. Intestinal transplantation with intestinal failure. Clin Perinatol 2013;40:161-174
Mark A. Arguments for routine administration of probiotics for NEC prevention. Curr Opin Pediatr 2019;31:188-194
Moore JE. Newer monitoring techniques to determine the risk of necrotizing enterocolitis. Clin Perinatol 2013;40:125-134
Nakayuenyongsuk W, Christofferson M, Stevenson DK, Sylvester K, et al. Point-of-care fecal calprotectin monitoring in preterm infants at risk for necrotizing enterocolitis. J Pediatr 2018;195:98-103
Neu J, Modi N, Caplan M. Necrotizing enterocolitis comes in different forms: historical perspectives and defining the disease. Semin Fetal Neonatal Med 2018;23:370-373
Neu J, Pammi M. Necrotizing enterocolitis: The intestinal microbiome, metabolome and inflammatory mediators. Semin Fetal Neonatal Med 2018;23:400-405
Ng PC, Chan KYY, Poon TCW. Biomarkers for prediction and diagnosis of necrotizing enterocolitis. Clin Perinatol 2013;40:149-159
Ng PC. An update on biomarkers of necrotizing enterocolitis. Semin Fetal Neonatal Med 2018;23:380-386
O´Neill Jr JA, Rowe MI, Grosfeld JL, et al (eds). Pediatric Surgery. Philadelphia: Elsevier, 2017
Pietz J, Achanti B, Lilien L, et al. Prevention of necrotizing enterocolitis in preterm infants: 20-year experience. Pediatrics 2007;119:e164- e170
Polin RA, Yoder MC. Workbook in Practical Neonatology. Philadelphia: Elsevier, 2015
Rao SC, Athalye-Jape GK, Deshpande GC, Simmer KN, Patole SK. Probiotic supplementation and late-onset sepsis in preterm infants: a meta-analysis. Pediatrics 2016;137 (3): e20163684
Reber K, Nankervis CA. Necrotizing enterocolitis: preventive strategies. Clin Perinatol 2004;31:157-167
Rocha G, Silva G, Virella D, Guimarães H. Enterocolite necrosante – Registo Nacional dos Recém-Nascidos de Muito Baixo Peso. Acta Pediatr Port 2003;34:153-157
Rose AT, Patel RM. A critical analysis of risk factors for necrotizing enterocolitis Semin Fetal Neonatal Med 2018;23:374-379
Saroha V, Josephson CD, Patel RM. Epidemiology of necrotizing enterocolitis. Clin Perinatol 2019;46:101-117. https://doi.org/10.1016/j.clp.2018.09.00
Seager E, Longley C, Aladangady N, et al. Measurement of gut oxygenation in the neonatal population using near-infrared spectroscopy: a clinical tool? Arch Dis Child Fetal Neonatal Ed 2020;105:F76–F86. doi:10.1136/archdischild-2018-316750
Shelby RD, Cromeens B, Rager TM, et al. Influence of growth factors on the development of necrotizing enterocolitis. Clin Perinatol 2019;46:51-64
Soll RF. Probiotics: are we ready for routine use? Pediatrics 2010;125:1071-1072
Szajewska H. Gut microbiota is a hot and fast‐moving topic, and paediatricians need to monitor the latest developments. Acta Paediatrica. 2019;108:1934-1935. DOI: 10.1111/apa.14960
Terrin G, Passariello A, De Curtis M, et al. Ranitidine is associated with infections, necrotizing enterocolitis, and fatal outcomes in newborns. Pediatrics 2012;129:e40– e45
Warner BB, Tarr PI. Necrotizing enterocolitis and preterm infant gut bacteria. Semin Fetal & Neonatal Med 2016;21:394e-e399
Warner BB, Deych E, Zhou Y, et al. Gut bacteria dysbiosis and necrotizing enterocolitis in very low birth weight infants: a prospective case-control study. Lancet 2016;387(10031):1928-1936
Wertheimer F, Arcinue R, Niklas V. Necrotizing Enterocolitis: enhancing awareness for the general practitioner. Pediatr Rev 2019;40:517-527. DOI: 10.1542/pir.2017-0338
Williams H. Perforation: how to spot free intraperitoneal air on abdominal radiograph. Arch Dis Child Educ Pract Ed 2006;91: e54- e57