Definição e importância do problema
A designação anomalias ano-rectais (AAR) engloba um conjunto diverso de defeitos congénitos gerados a partir da 5ª a 8ª semanas de gestação, incluindo o ânus imperfurado e suas variantes. O espectro de manifestações clínicas é também muito diverso, desde formas clínicas de gravidade diminuta a formas clínicas extremamente complexas e graves.
As anomalias ano-rectais, fazendo parte das síndromas de defeito de regressão caudal, surgem com uma frequência de cerca de 1/5.000 nascimentos, com predomínio no sexo masculino; as formas menos complexas verificam-se no sexo feminino.
Sistematização anatómica
No sexo masculino as anomalias ano-rectais são sistematizadas, por ordem crescente de complexidade, do seguinte modo:
- AAR com ânus coberto (cobertura cutânea membranosa simples do orifício anal);
- AAR com fístula perineal (trajecto fistuloso para a pele perineal ou rafe mediana escrotal);
- AAR com atrésia rectal (atrésia da continuidade ano-rectal após a linha pectínea, com permanência do segmento cólico distal);
- AAR com fístula para o aparelho urinário (contacto entre o aparelho digestivo e o aparelho urinário ao nível da uretra bulbar, prostática ou colo da bexiga).
No sexo feminino, as referidas anomalias podem apresentar-se clinicamente, também por ordem crescente de complexidade, do seguinte modo:
- AAR com ânus coberto (cobertura cutânea simples do orifício anal);
- AAR com fístula perineal (trajecto fistuloso para a pele perineal);
- AAR com fístula vestibular (trajecto fistuloso para a forchette vaginal);
- AAR com fístula vaginal (trajecto fistuloso para a parede posterior da vagina retro-himenal);
- AAR com formação de cloaca (orifício perineal único, hipoplasia dos genitais externos, ausência de orifício anal, canal comum e sinus urogenital).
Manifestações clínicas e diagnóstico
O dado clínico fundamental desta anomalia é a ausência de orifício anal de forma e localização normal, o que é comprovado no âmbito do primeiro exame clínico sistemático do recém-nascido no pós-parto imediato.
No sexo masculino, pode ser acompanhada de períneo mal desenvolvido com sulco internadegueiro não proeminente, e presença de fístula para o aparelho urinário em cerca de 90% dos casos. No sexo feminino podem ser observados: presença de fístula para a pele perineal ou para a vagina, ou ainda, orifício perineal único, acompanhado de hipoplasia genital marcada.
As anomalias congénitas do aparelho urinário acompanham as anomalias ano-rectais em cerca de 48% dos casos. Outras anomalias frequentemente associadas incluem as cardíacas, digestivas e vertebrais (hemivértebras, disrafismo e agenésia sagrada). Associações possíveis de defeitos acompanhantes incluem as designadas pelas siglas VACTERL (defeitos: vertebral, anal, cardíaco, traqueal, esofágico, renal, membro/limb), e VATERR (defeitos: vertebral, anal, traqueal, esofágico, radial, renal). Como regra geral pode estabelecer-se que as anomalias associadas constituem o factor prognóstico mais importante das anomalias ano-rectais (por ex., boa correlação entre o grau de desenvolvimento do sacro e a futura função: ausência de sacro associa-se a incontinência fecal e urinária).
O diagnóstico de anomalia ano-rectal é fundamentalmente clínico. Os exames perineal e genital fornecem o diagnóstico, permitindo definir, na grande maioria das vezes, o tipo anatómico em causa.
Nos casos de exame clínico detectando “ânus imperfurado”, para avaliação da distância entre a solução de continuidade rectal e a superfície cutânea sem orifício anal, era clássico até há 2 décadas realizar radiografia abdominal simples colocando placa radiopaca de chumbo sobre o períneo (região anal). (Figura 1)
Decorrendo da probabilidade de associação doutras anomalias, como atrás foi referido, estão indicados os seguintes exames complementares:
estudo radiológico sumário da coluna dorso-lombossagrada (em dois planos); estudo ecográfico da coluna lombar para diagnóstico precoce de síndroma de medula ancorada ou de regressão caudal; ecografia do aparelho urinário, extremamente importante para o diagnóstico imediato de qualquer defeito estrutural renal e do aparelho excretor. Para o diagnóstico de defeitos de encerramento do tubo neural e em situações específicas poderá haver necessidade de RM.
Tratamento
As AAR têm sempre indicação operatória. O fundamento da intervenção cirúrgica é a criação de um orifício anal bem posicionado anatomicamente, normofuncionante e completamente separado do aparelho urinário e do aparelho genital.
A decisão terapêutica imediata mais importante prende-se com a eventual necessidade de construção de uma colostomia diversiva, no quadrante inferior esquerdo, utilizando um segmento de junção entre o cólon descendente e o cólon sigmoideu. Esta decisão terapêutica depende da definição anatómica e diagnóstica do tipo de anomalia ano-rectal, e deverá ser tomada após um intervalo de 16 a 24 horas depois do nascimento; com efeito, verificando-se neste período a progressão da massa meconial até zona mais distal do tubo digestivo, e a possibilidade de preenchimento de eventual trajecto fistuloso cutâneo existente, é possível tirar conclusões mais definitivas quanto à modalidade de tratamento.
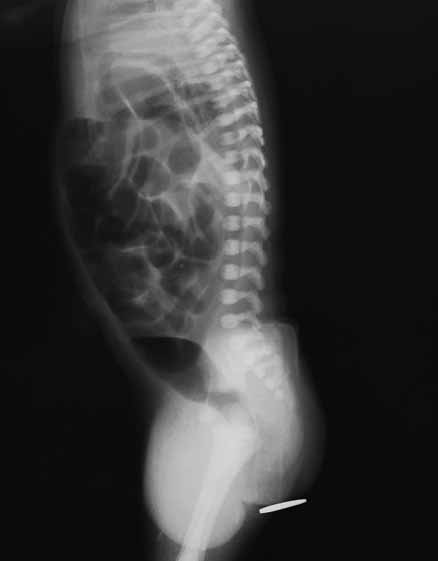
FIGURA 1. RN com ânus imperfurado (placa de chumbo colocada na região anal para avaliar a distância entre a pele e a zona de interrupção anómala rectal (ausência de imagens gasosas). Actualmente, através da ecografia pode determinar-se o local onde se verifica interrupção do trânsito (Imagem radiográfica com interesse histórico).
Este aspecto é de extrema importância, porque um defeito não complexo, não necessitando de colostomia diversiva, poderá ser corrigido definitivamente no período neonatal. Pelo contrário, as anomalias mais complexas necessitam de colostomia diversiva para evitar a retenção fecal e a dilatação distal da bolsa cólica, com riscos de perfuração, de infecção urinária (que se pode complicar por um quadro de sépsis urinária) e de reabsorção de urina pela mucosa intestinal conducente a acidose metabólica. Estas situações constituem, efectivamente, risco de vida para qualquer recém-nascido afectado por uma forma complexa de anomalia ano-rectal com fístula recto-urinária.
Reportando-nos à sistematização anatómica descrita noutra alínea, referem-se agora as variedades anatómicas necessitando de colostomia.
No sexo masculino:
- AAR com atrésia rectal;
- AAR com fístula para o aparelho urinário.
No sexo feminino:
- AAR com fístula vestibular;
- AAR com fístula vaginal;
- AAR com formação de cloaca.
Notas importantes:
|
Complicações pós-operatórias
As complicações resultam das múltiplas intervenções cirúrgicas a que os doentes com esta patologia são submetidos. As mais importantes são: deiscências de vários tipos, estenose, prolapso da mucosa cólica, má posição da anoplastia em relação aos limites do complexo muscular perineal e refistulização recto-urinária ou recto-vaginal.
Seguimento
Os doentes portadores de AAR, nomeadamente nas suas formas mais complexas, necessitam de um seguimento multidisciplinar em regime ambulatório, englobando designadamente diversas áreas como: enfermagem, fisioterapia, fisiatria, cirurgia pediátrica, pediatria médica, medicina familiar, nefrologia pediátrica, neurologia pediátrica e ginecologia pediátrica.
As situações clínicas mais problemáticas associadas ao seguimento da AAR são a obstipação pós-operatória, a incontinência fecal e a incontinência urinária. A obstipação pós-operatória é a sequela mais comum dos doentes com AAR. Paradoxalmente, é mais grave nas formas clínicas mais ligeiras. A obstipação deverá ser tratada agressivamente e de modo prolongado para evitar as suas consequências nefastas, como o megarrecto. A incontinência fecal, que pode surgir em cerca de 30% de todos os casos, implica a necessidade de programa de reeducação intestinal adaptado a cada doente. O seu fundamento é a utilização criteriosa e individualizada de laxantes, emolientes e clisteres de limpeza.
Prognóstico
O prognóstico dos doentes com AAR depende, não só do sucesso do acto cirúrgico a que foram submetidos, mas, principalmente, do tipo de AAR e da patologia subjacente e acompanhante da mesma.
Reiterando o que foi descrito anteriormente, os factores prognósticos fundamentais são: a evolução do status nefrológico e a capacidade de continência esfincteriana vesical e anal. Estes dois aspectos são os mais importantes na definição da qualidade de vida futura destes doentes.
BIBLIOGRAFIA
Ashcraft KW, Holcomb GW III, Murphy JP (eds). Pediatric Surgery. Philadelphia: Saunders, 2005
Cloherty JP, Eichenwald EC, Strak AR. Manual of Neonatal Care. Philadelphia; Lippincott Williams & Wilkins, 2008
Coran AG. Pediatric Surgery. Philadelphia: Elsevier, 2013
Garcia JJ, Cruz O, Mintegi S, Moreno JM (eds). M Cruz Manual de Pediatria. Madrid: Ergon, 2020
Guimarães JC, Carneiro MJ, Loio P, Macedo A, Tuna ML, et al. Manual Prático de Neonatologia. Lisboa: Hospital de S. Francisco Xavier/Uriage, 2016
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
O´Neill Jr JA, Rowe MI, Grosfeld JL, et al (eds). Pediatric Surgery. Philadelphia: Elsevier, 2017
Peña A, Guardino K, Tovilla J. Bowel management for fecal incontinence in patients with anorectal malformations. J Pediatr Surg 1998; 33: 133-137
Peña A, Hong AR. Advances in the management of anorectal malformations. Am J Surg 2000; 180: 370-376
Polin RA, Yoder MC. Workbook in Practical Neonatology. Philadelphia: Elsevier Saunders, 2015
Polin RA, Abman SH, Rowitch DH, Benitz WE, Fox WW (eds). Fetal and Neonatal Physiology. Philadelphia: Elsevier, 2017
Rosen R, Buonomo C, Andrade R, Nurko S. Incidence of spinal cord lesions in patients with intractable constipation. J Pediatr 2004; 145: 409-411
Wyllie R, Hyams JS, Kay M (eds). Pediatric Gastrointestinal and Liver Disease. Philadelphia: Elsevier, 2016