Importância do problema
A obstrução do tracto urinário pode ser congénita (anatómica), ou adquirida, isto é, causada por traumatismo, neoplasia, cálculos, processos inflamatórios e, finalmente, como complicação de procedimentos cirúrgicos.
Na maior parte dos casos, trata-se de lesões de natureza congénita.
As lesões obstrutivas podem localizar-se a qualquer nível, desde o meato uretral às infundíbula caliciais.
As repercussões sobre o parênquina renal dependem do nível da obstrução, do seu grau, da idade da criança e do modo como surge (agudo ou crónico).
Seguidamente são descritas algumas das entidades clínicas mais representativas deste tipo de patologia.
SÍNDROMA DA JUNÇÃO PIELO-URETERAL
Definição e aspectos epidemiológicos
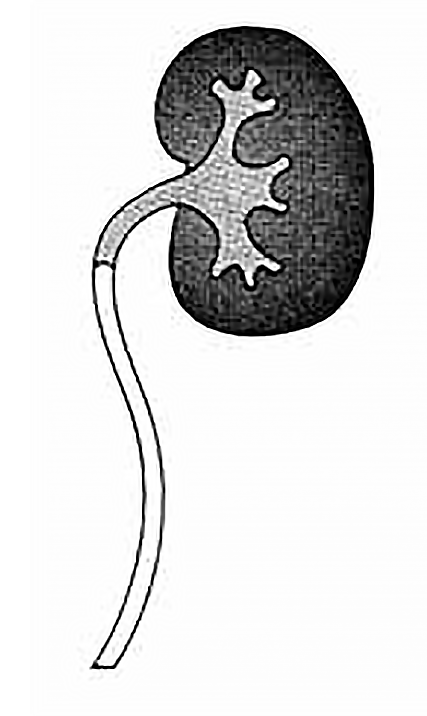
A síndroma da junção pielo-ureteral (SJPU) é uma anomalia do aparelho urinário superior caracterizada pela obstrução funcional ou estrutural da junção pielo-ureteral, de carácter completo ou parcial. Essa obstrução pode ser devida a um obstáculo mucoso endoluminal do uréter, a disposição anómala das fibras espirais da parede ureteral, a válvula anómala do uréter, a angulação do mesmo, a compressão extrínseca por bandas fibróticas retroperitoneais, ou por vaso anómalo originário da artéria renal polar inferior.
A SJPU é responsável por dilatação progressiva do bacinete e dos cálices renais e, em estádios mais avançados, por compressão do parênquima renal com alterações da diferenciação córtico-medular e parênquimo-sinusal.
Este defeito tem uma incidência de 1/2.000 nascimentos, sendo mais frequente no sexo masculino e no lado esquerdo. A anomalia pode ser bilateral em 20% dos casos.
Manifestações clínicas e diagnóstico
Actualmente, devido aos progressos verificados na vigilância pré-natal e à rotina do estudo ecográfico durante o referido período, esta anomalia do aparelho urinário é a mais frequentemente detectada por diagnóstico pré-natal.
De salientar que o achado ecográfico de dilatação do excretor superior não é sinónimo ou indicativo de obstrução mecânica total, podendo esta situação desaparecer no período neonatal precoce; contudo, tal achado obriga a uma atitude de observação clínica e imagiológica seriada.
Os factores de decisão quanto à atitude mais correcta a tomar são: a sintomatologia, a evidência de agravamento imagiológico, e a degradação progressiva da função renal.
Por essa razão, está indicado proceder a um conjunto de exames complementares que nos auxiliam na decisão terapêutica durante o período neonatal.
Nos doentes sem diagnóstico pré-natal ou sem sintomatologia neonatal, o quadro clínico pode ser muito variável. Pode manifestar-se como infecção urinária, como dor abdominal – lombar recorrente, como massa lombar pela dilatação extrema do bacinete, ou como hipertensão arterial.
Na segunda infância, a SJPU pode ser causa de hematúria macroscópica pós-traumatismo do rim, mesmo ligeiro.
O diagnóstico deste defeito é fundamentalmente imagiológico. A ecografia permite identificar a dilatação do bacinete e dos cálices renais, e também estabelecer a diferenciação córtico-medular e parênquimo-sinusal no rim afectado.
O renograma isotópico DTPA permite confirmar a presença de obstrução mecânica à excreção do radiofármaco, mais evidente na prova com furosemido. Salienta-se que estes exames podem ser unicamente realizados após a 3ª ou 4ª semana de vida por limitações técnicas que se prendem com o peso do recém-nascido, a sua capacidade de processamento do radiofármaco e a captação pelos aparelhos de leitura.
Actualmente, o uso de renograma com MAG3 pode encurtar o intervalo de estudo para a 2ª semana de vida. Com efeito, o renograma isotópico permite avaliar a função renal diferencial e a taxa de filtração glomerular.
A utilização de contraste iodado convencional na urografia endovenosa (de eliminação) está reservada apenas para casos particulares para delimitação anatómica (por imagem) do excretor alto.
Indicação cirúrgica e terapêutica operatória
Os critérios de indicação cirúrgica na SJPU são de carácter clínico e funcional.
Os critérios clínicos são:
- Existência de massa renal palpável;
- Sintomatologia dolorosa; ou
- Infecção renal recorrente ou de carácter subclínico.
Os critérios funcionais são:
- Valor da medição ecográfica do bacinete superior a 20 mm;
- O atraso de excreção de radiofármaco superior a 20 minutos; e
- Função renal diferencial (FRD) inferior a 35% (do lado afectado em relação ao contralateral).
As várias opções cirúrgicas disponíveis para a resolução do SJPU baseiam-se no princípio da ressecção da zona estenosada e ulterior anastomose alargada ao bacinete, permitindo uma drenagem passiva mais eficaz. A pieloplastia de Anderson-Haynes é a técnica cirúrgica mais utilizada na maior parte dos casos.
Complicações pós-operatórias
A pieloplastia é um procedimento cirúrgico com uma frequência baixa de complicações. As complicações pós-operatórias mais descritas estão associadas a infecção da ferida operatória e à pequena extravasão de urina pela anastomose pielo-ureteral.
Seguimento
O seguimento da situação é realizado em ambulatório com a realização de ecografia ao 3º mês e renograma de controlo ao 6º mês pós-operarório. Até então deverá ser mantida a profilaxia antimicrobiana iniciada, uma vez estabelecido o diagnóstico da situação. Sobre a profilaxia antimicrobiana existe controvérsia, não sendo utilizada em todos os centros.
Prognóstico
Na ausência de complicações cirúrgicas, nomeadamente urinoma pós- operatório ou estenose da anastomose pielo-ureteral, o prognóstico é em geral bom, tendo a cirurgia correctiva sucesso em mais de 95% dos casos.
MEGAURÉTER OBSTRUTIVO
Definição e aspectos epidemiológicos
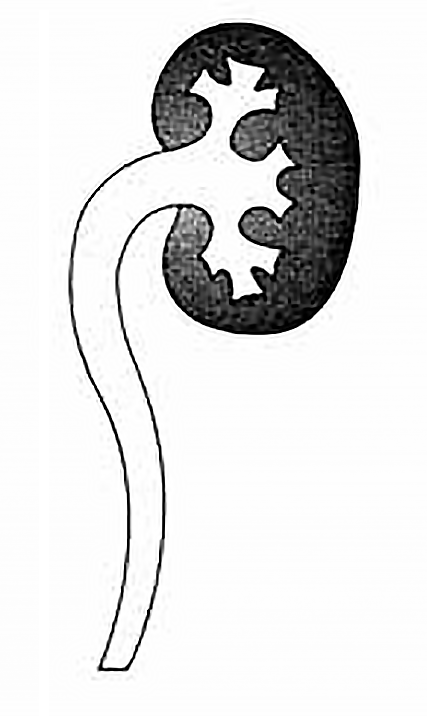
O megauréter é uma anomalia funcional do uréter distal que dificulta a passagem da urina para a bexiga, de tal resultando dilatação a montante associada na maioria dos casos a dilatação pielocalicial.
Especificamente, o conceito de megauréter obstrutivo diz respeito à dilatação do uréter (> 7 mm) com aumento da pressão intraluminal, de natureza congénita obstrutiva, surgindo como resultado de estenose da junção ureterovesical, ou de implantação ectópica do mesmo. A incidência é cerca de 1/2.000.
O megauréter pode ser bilateral em cerca de 25% a 30% dos casos; o mesmo defeito, na grande maioria, está associado a duplicidade da árvore excretora.
Etiopatogénese
Esta anomalia é devida a uma alteração histológica da porção terminal do uréter com hipertrofia das fibras de colagénio, escassez de fibras musculares longitudinais e hiperplasia de fibras musculares circulares.
Manifestações clínicas e diagnóstico
Esta situação tem diagnóstico pré-natal frequente (cerca de 65%), sendo característica do sexo masculino.
O quadro clínico, relativamente inespecífico, revela-se, na maior parte dos casos por infecção urinária recorrente.
A litíase ureteral pode também complicar o quadro obstrutivo em cerca de 5% dos casos. A existência de um megauréter obstrutivo agravado pelo encravamento distal de cálculos constitui uma urgência em urologia, tendo indicação para descompressão imediata.
Como exames complementares de diagnóstico, a ecografia renal, vesical e ureteral fornecem uma imagem de uréter-hidronefrose volumosa, muitas vezes associada a alterações da diferenciação córtico-medular e parênquimo-sinusal renal.
A cistografia com tempo miccional deverá ser realizada para exclusão da etiologia refluxiva do megauréter.
O renograma isotópico com DTPA ou MAG3 confirma a natureza obstrutiva da lesão e permite avaliar a função renal diferencial. A cintigrafia renal com DMSA faculta a imagem da perfusão do parênquima e a possível presença de cicatrizes renais pós-infecciosas. A urografia endovenosa permite realizar a definição anatómica da porção distal do uréter, e da sua relação com a junção uréter-vesical, ou da sua inserção ectópica.
Indicação cirúrgica e terapêutica operatória
A indicação cirúrgica desta situação é essencialmente colocada na presença de defeito anatómico da junção uréter-vesical e de implantação anómala do uréter ectópico no colo vesical. Outra indicação cirúrgica premente é a degradação progressiva da função renal, secundária à presença de infecção urinária recorrente associada a esta anomalia.
A terapêutica cirúrgica baseia-se na reimplantação uréter-vesical eutópica (ou no local normal) e na ureteroplastia de redução de calibre quando necessária, para se poder reimplantar, com segurança, o uréter na bexiga.
Complicações pós-operatórias
As complicações pós-operatórias mais frequentes são a manutenção do quadro obstrutivo e a persistência de infecção urinária recorrente. Poderá também haver desenvolvimento de um quadro refluxivo, por ostium ureteral reimplantado com função anómala.
Seguimento
Importa salientar que as imagens de dilatação do uréter se mantêm na ecografia e na radiologia convencional mesmo após cirurgia de reimplantação com sucesso. Efectivamente, a dilatação ureteral no seu trajecto lombar e pélvico não é alterada com a intervenção cirúrgica; tal facto não deve ser considerado alarmante, excepto nos casos de manutenção do quadro de infecção urinária ou de dor lombar por distensão da árvore excretora.
O megauréter obstrutivo implica acompanhamento assíduo por equipa multidisciplinar (pediatra, imagiologista, etc.) pelo risco de degradação progressiva da função renal.
O seguimento imagiológico pós-operatório é realizado por meio de ecografia renal ao terceiro mês, e por renograma isotópico ao sexto mês. Posteriormente, é necessário realizar um renograma isotópico com uma periodicidade anual em simultâneo com estudo de parâmetros laboratoriais da função renal.
Prognóstico
O prognóstico desta patologia depende directamente do status da função renal pré-operatória. A correcção cirúrgica do megauréter, quando realizada com sucesso, pode evitar a degradação progressiva da função renal, mas não pode melhorar o status renal decorrente da situação pré-operatória.
Na ausência de complicações pós-operatórias e de degradação progressiva da função renal o prognóstico é, em geral, bom.
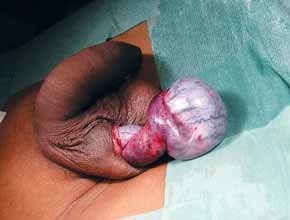
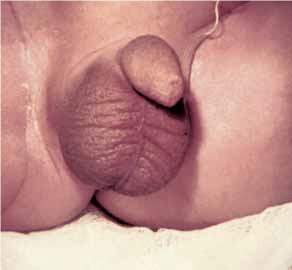
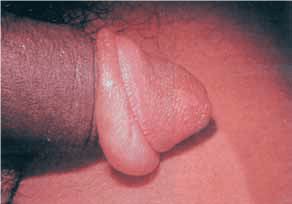
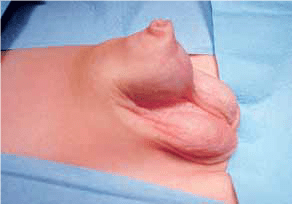
URETEROCELE
Definição
O ureterocele é uma dilatação quística terminal da porção intravesical do uréter, geralmente associada a estenose do ostium ureteral. Esta anomalia está associada a duplicidade do sistema excretor em 80% dos casos (pielão superior) e a ectopia e bilateralidade, respectivamente em 60% e 10% dos casos.
Manifestações clínicas e diagnóstico
Actualmente, cerca de 60% dos casos de ureterocele têm diagnóstico pré-natal. A clínica é muito pouco característica e manifesta-se fundamentalmente por infecção urinária. No sexo feminino pode manifestar-se por incontinência urinária primária permanente.
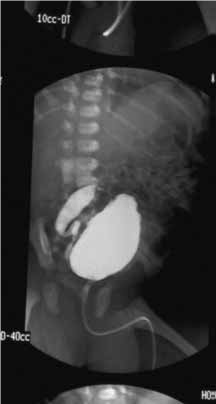
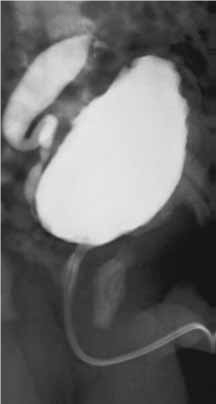
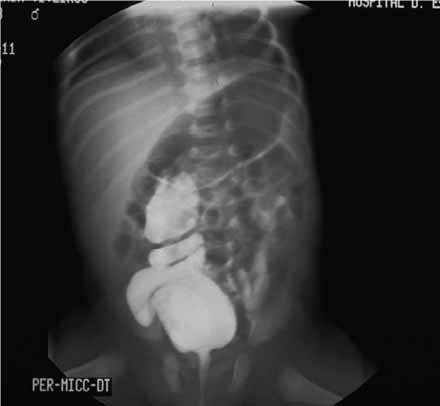
O diagnóstico é feito, na maior parte dos casos, por meio ecográfico, no âmbito da investigação de um quadro de infecção urinária. A ecografia fornece imagens características compatíveis com a presença de uma massa quística intravesical, com uréter pélvico visível. O exame ecográfico permite também diagnosticar a presença de sistemas duplos de drenagem renal. A urografia de eliminação permite delinear a anatomia do sistema excretor, a ectopia da relação distal do uréter, e a típica imagem translúcida de subtracção intravesical que é patognomónica. A cistouretrografia com tempo miccional permite diagnosticar a presença de refluxo para o pielão inferior nos sistemas duplos (50% dos casos), e para o uréter contralateral (25% dos casos).
Indicação operatória e terapêutica cirúrgica
O ureterocele tem sempre indicação cirúrgica, porque: – está associado a alterações anatómicas e funcionais da relação distal do uréter; e – cursa frequentemente com um quadro de infecção urinária recorrente.
O objectivo do tratamento do ureterocele é o controlo da infecção urinária, a preservação da função renal, a protecção funcional das unidades de drenagem renal normal homolaterais ou contralaterais, e a manutenção da continência urinária.
Nos ureteroceles pequenos, eutópicos e não associados a dilatação ureteral de grande dimensão, o tratamento cirúrgico pode ser realizado por via endoscópica transuretral com ressecção do mesmo. Este tipo de tratamento tem sucesso clínico em cerca de 85% dos casos; a descompressão do ureterocele está especialmente indicada no recém-nascido. Se houver RVU secundário ao procedimento, a reimplantação secundária é necessária.
Nos casos de ureteroceles de grande dimensão, associados a duplicidade da árvore excretora renal ou ectópicos, a terapêutica cirúrgica convencional é a mais indicada. Nesta abordagem cirúrgica, por incisão lombar, deverá ser feita a ressecção do pielão superior renal, com excisão do megauréter; e, por incisão vesical, a excisão do ureterocele, e reimplantação eutópica do uréter originário do pielão inferior no trígono vesical homolateral.
Complicações pós-operatórias
A complicação pós-operatória mais frequente é o estabelecimento de RVU secundário, quer ao procedimento endoscópico, quer à terapêutica cirúrgica. Ambas as situações obrigam a correcção ulterior por cirurgia anti-refluxiva. Por vezes, poderá haver lesões iatrogénicas do colo vesical secundárias à ressecção de ureteroceles gigantes interessando a região do colo, com consequente perturbação dos mecanismos de continência do colo.
Seguimento
O seguimento deve ser rigoroso e estar focado no controlo e preservação da função renal bilateral, assim como na manutenção dos mecanismos de continência urinária.
Estes doentes necessitam:
- de controlo ecográfico na terceira semana pós-operatória para certificação do sucesso cirúrgico da ressecção do ureterocele;
- da realização de cistografia miccional para rastrear o estabelecimento de RVU pós-procedimento; e
- de estudo isotópico anual para controlo da função renal, total e diferencial.
Prognóstico
O prognóstico global depende da função renal residual. A terapêutica cirúrgica é correctiva em mais de 90% dos casos.
VÁLVULAS DA URETRA POSTERIOR
Importância do problema
As válvulas da uretra posterior (VUP) são pequenas pregas mucosas da uretra masculina, que se constituem como obstáculo ao fluxo anterógrado e normal de urina.
Estão localizadas na crista uretral, na proximidade do veru montanum. Tal anomalia já existe por volta da 12ª semana gestacional, quando começa a formar-se urina.
Considerando a globalidade dos processos obstrutivos infravesicais, as VUP constituem as situações de maior relevância, uma vez que o grau e duração da obstrução poderá conduzir a lesão renal irreversível na ausência de tratamento.
Quanto à localização, estas anomalias são classificadas do seguinte modo: tipo I (as mais frequentes e mais distais ao veru montanum); tipo II (muito raras, entre o colo da bexiga e o veru montanum) e tipo III (sobre o veru montanum).
Actualmente, as VUP integram-se na síndroma denominada síndroma válvula-bexiga, à qual está ligada um conceito funcional: a persistência de obstrução uretral origina disfunção vesical grave (bexiga permanecendo com baixa capacidade em repouso e elevadas pressões sob enchimento) e ulterior desenvolvimento de uréter-hidronefrose ascendente com repercussão funcional renal.
Manifestações clínicas e diagnóstico
As manifestações clínicas deverão ser consideradas dentro de um espectro de gravidade variável.
As formas mais graves manifestam-se já no recém-nascido. Por outro lado, existem casos de obstrução discreta, assintomáticos durante um período longo de tempo.
Simultaneamente, existem formas de apresentação de extrema gravidade, de expressão ecográfica pré-natal com oligoâmnio, sinais de dilatação do aparelho excretor alto e alterações do normal desenvolvimento do parênquima renal bilateral.
As manifestações clássicas, já detectáveis no recém-nascido, são caracterizadas por jacto urinário fraco ou gotejante, sinais de retenção vesical (saliência hipogástrica dura e não depressível relacionada com bexiga de parede espessada e com hipertrofia muscular da mesma), massa abdominal-lombar compatível com mega-uréteres refluxivos, e hidronefrose marcada do excretor alto. Por vezes, por perfuração do tracto excretor, pode surgir ascite urinária volumosa.
A infecção urinária resultante da retenção grave pode ser complicada de sépsis urinária. As VUP podem igualmente ser causa de hipertensão arterial no recém-nascido.
O diagnóstico da situação é, por conseguinte, feito em função da clínica e dos exames complementares.
O exame ecográfico pós-natal pode oferecer imagem de uma bexiga de parede espessada por hipertrofia muscular, com trabeculação da mucosa, existência de divertículo da bexiga e dilatação do tracto excretor por refluxo vésico-ureteral de grau elevado. Pode verificar-se também a presença de hidronefrose do excretor alto. Dependendo do compromisso do desenvolvimento do parênquima renal, poderão ser evidentes imagens de displasia renal bilateral.
O exame de excelência ou padrão de ouro para o diagnóstico de VUP é a cistografia miccional. Este exame permite, na fase de enchimento vesical, quantificar a capacidade da bexiga, delimitar o contorno mucoso da mesma (presença de trabeculações), detectar a presença de divertículos e de refluxo vésico-ureteral; e, na fase miccional, evidenciar as alterações típicas da uretra posterior na presença de válvulas da uretra: alongamento e alargamento da uretra prostática e hipertrofia do colo da bexiga.
O exame que permite a definição anatómica de VUP é a uretrocistoscopia. Com esta técnica é possível obter a imagem em tempo real do tipo específico de VUP, assim como a sua localização em relação ao veru montanum. Por outro lado, com a mesma, é possível a terapêutica das VUP por ablação endoscópica. Esta manobra cirúrgica pode ser realizada no período neonatal.
Nos casos de incapacidade técnica de ablação endoscópica (por ex. RN de muito baixo peso), poderá ser necessário construir uma vesicostomia temporária para derivar o tracto urinário pré-uretral.
Prognóstico
As VUP são uma situação clínica cujo prognóstico decorre do espectro de apresentação e da precocidade da ablação cirúrgica.
Com efeito, o tratamento endoscópico precoce evita o agravamento progressivo da disfunção vesical e do refluxo vésico-ureteral de grau elevado, o que contribui para o não agravamento do status funcional renal. O estádio mais grave é caracterizado por displasia renal bilateral, com episódios de hipertensão e insuficiência renal de instalação progressiva (a qual poderá surgir com frequências oscilando entre 25 e 50%). (ver no capítulo sobre RVU – síndroma de Mitchel: válvula-bexiga-rim)
Inversamente, a evicção do obstáculo ao esvaziamento vesical promove o crescimento harmónico e cíclico da bexiga, contribuindo para uma capacidade normal, com pressões de enchimento e de esvazimento também normais. De igual modo, a inexistência de refluxo vésico-ureteral e de uretér-hidronefrose volumosa, evita a degradação renal progressiva, promovendo o desenvolvimento e diferenciação do rim do recém-nascido.
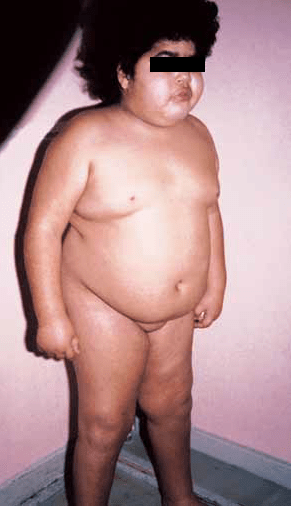
SÍNDROMA DE EAGLE BARRETT (prune-belly)
No âmbito da abordagem do tópico “Obstrução do tracto urinário” é clássico mencionar esta situação rara (1/40.000 RN) sendo 95% do sexo masculino, com elevada mortalidade fetal. Esta síndroma caracteriza-se fundamentalmente por deficiente desenvolvimento da musculatura abdominal e da bexiga, aspecto flácido e pregueado da pele abdominal (daí o nome de “barriga com aspecto de abrunho”), e obstrução do tracto urinário incluindo uretra. De tal resultam oligoâmnio, por vezes ascite urinária, fácies Potter e hipoplasia pulmonar. Outras anomalias incluem displasia renal, defeitos cardíacos, ectopia testicular, etc..
O prognóstico depende da hipoplasia pulmonar e do grau de disfunção renal. Há casos submetidos a transplante renal com bons resultados.