DEFINIÇÃO E IMPORTÂNCIA DO PROBLEMA
A doença de Kawasaki (DK), também anteriormente denominada síndroma linfomucocutânea, foi descrita pela primeira vez no Japão por Tomikasu Kawasaki, em 1967. Define-se como uma vasculite necrosante sistémica aguda, afectando predominantemente vasos de médio calibre, mas também, arteríolas, vénulas e capilares, com predilecção para as artérias coronárias. Tal nosologia comporta, pois, risco elevado de isquémia e de enfarte do miocárdio.
Constitui a vasculite aguda sistémica mais frequente na criança e a principal causa de cardiopatia adquirida nos países desenvolvidos.
ASPECTOS EPIDEMIOLÓGICOS
Na grande maioria dos casos (cerca de 80%) ocorre abaixo dos 5 anos de idade, sendo rara antes dos 6 meses, e muito rara depois dos 5 anos. A explicação provável para esta distribuição assenta na imunidade passiva antes dos 6 meses (anticorpos maternos transplacentares) e na imunidade adquirida depois dos 5 anos; contudo há casos descritos no recém-nascido.
O sexo masculino é mais frequentemente afectado na proporção aproximada de 2/1.
De acordo com estudos epidemiológicos parece tratar-se duma doença sazonal com “pico” de incidência no Inverno e Primavera. A incidência, máxima na Ásia e, designadamente, no Japão, (90/100.000) tem vindo a aumentar, situando-se actualmente entre 3-5/100.000 no Reino Unido e 6-11/100.000 nos EUA, Canadá e em Portugal. É provável que tal aumento se relacione com uma maior possibilidade diagnóstica, decorrente do melhor conhecimento da doença. Parece existir predisposição genética (nomeadamente em relação com a existência de genes determinantes dos seguintes tipos no sistema HLA: B5, B44, Bw51, DR3 e DRB3*0301). A recorrência é rara. Estudos genéticos identificaram casos com elevada susceptibilidade de receptores de citocinas relacionados com genes CCL3 e CCL3L1, e em portadores de variantes de certos genes tais como o CCR5.
ETIOPATOGÉNESE
A etiopatogénese da DK é ainda desconhecida. Todavia, admite-se a hipótese de comparticipação muito provável de germes microbianos tendo em conta, nomeadamente: a semelhança das manifestações clínicas com certas doenças infecciosas como a escarlatina e a síndroma de choque tóxico, a sazonalidade em muitas áreas geográficas, a ocorrência de surtos, o risco aumentado de doença após contacto com outros casos, a maior frequência entre os 6 meses e os 5 anos, e a eficácia terapêutica da imunoglobulina. Contudo, não se conseguiu, até hoje, o isolamento de qualquer microrganismo responsável por esta entidade nosológica.
Determinados estudos identificaram, em casos fatais, antigénio associado à DK em corpos de inclusão de células epiteliais ciliadas brônquicas, admitindo-se: 1) que tais corpos de inclusão correspondem a agregados de proteínas víricas; e 2) que a porta de entrada do “possível agente causal” seja a via respiratória.
Noutros estudos é sugerido que as profundas anomalias imunorreguladoras encontradas na DK se devem a toxinas bacterianas (e/ou víricas) proteicas, capazes de se ligarem simultaneamente aos receptores das células T e às moléculas do MHC* de classe II, em zona deslocada ou fora do habitual sulco peptídico de união. Desta forma, este “desvio”, à margem do “sistema chave-fechadura”, permite que as referidas toxinas actuem de forma inespecífica em zonas mais amplas e em grande escala, sendo, por isso, designadas por superantigénios.
Tais superantigénios diferem dos antigénios convencionais em vários aspectos importantes: 1) activação policlonal das células B; 2) estimulação de um número maciço de linfócitos T circulantes capazes de se ligarem a um receptor de superfície celular específico; e 3) produção intensiva de citocinas pró-inflamatórias.
*A propósito do chamado CMH (ou MHC) – sigla em português de Complexo Major de Histocompatibilidade – recorda-se que existe um locus no braço curto do cromossoma 6 compreendendo múltiplos genes que determinam os antigénios (glicoproteínas de superfície) de histocompatibilidade (HLA ou human leucocyte antigens) de diversas características ou classes designadas por I, II e III, os quais desempenham papel importante nas interacções entre células do sistema imunitário; os antigénios de classe II são reconhecidos por células CD4. |
Apenas um número limitado de linfócitos responde a um antigénio convencional (tipicamente < 1 célula em cada 10.000 linfócitos). Pelo contrário, os superantigénios podem activar até 20 a 30% dos linfócitos T circulantes, com subsequente libertação de quantidades extremamente elevadas de citocinas; estas últimas posteriormente dão origem a uma cascata de eventos traduzindo activação imunológica de grau extremo, a qual pode ser tipificada pela elevação sérica de todas as imunoglobulinas.
A febre alta e elevação dos reagentes de fase aguda podem ser secundárias ao aumento de IL-1, IL-6 e de TNF-α. O processo adenopático cervical pode reflectir a activação marcada das células B e T.
A lesão vascular em geral, assim como a lesão do endotélio das coronárias pode resultar: 1) da activação de células endoteliais e de moléculas de adesão leucocitária; 2) da infiltração de células CD4+ e CD8+, assim como de macrófagos; 3) da resposta pró-inflamatória e pró-trombótica exagerada produzida pelo excesso de citocinas; e 4) da acção de neoantigénios sobre o referido endotélio coronário.
Todos estes mecanismos possíveis ocorrem seguramente em indivíduos geneticamente predispostos, esta contribuição importante é suportada pela manutenção do elevado grau de incidência em indivíduos asiáticos mesmo que habitem em países com menor incidência. Também estudos recentes de identificação fenotípica demonstraram polimorfismos de genes que se associam a aumento da incidência de lesão coronária, da sua gravidade, recorrência da doença e até resistência ao tratamento com imunoglobulinas.**
**Foram recentemente identificados genes que determinam maior susceptibilidade para a DK e maior risco de complicações: o gene Inositol 1,4,5-triphosphate 3-kinase C – ITPKC ITPKC, é um regulador negativo da activação dos linfócitos T, os pacientes com DK teriam redução da actividade do ITPKC com consequente resposta inflamatória exagerada mediada por células T; polimorfismos do cluster de genes CCR3-CCR2-CCR5 determinam a resistência ao tratamento com IGIV e dessa forma maior gravidade da doença; na presença dos alelos HH e HR (Taniuchi et al – Genotipos de Fcg RIIIb-NA(1,2), Fcg RIIa-H/R131, and Fcg RIIIa-F/V158) existe maior propensão para o desenvolvimento de aneurismas coronários. |
É plausível o seguinte mecanismo fisiopatológico, descrito por fases: 1) num indivíduo geneticamente predisposto; 2) determinado germe microbiano produtor de superantigénios coloniza as membranas mucosas do tracto gastrintestinal; 3) a toxina é absorvida através da mucosa inflamada, estimulando as células mononucleares locais e/ou circulantes a produzir citocinas pró-inflamatórias que, por sua vez, provocam a febre e as restantes alterações clínicas observadas na DK; 4) em resposta à estimulação induzida por estes mediadores químicos, neoantigénios actuando no endotélio, tornam este especialmente susceptível à agressão por anticorpos citotóxicos e por células T activadas; 5) verifica-se, então, edema endotelial e do músculo liso vascular, do que resulta extensão do infiltrado inflamatório às restantes camadas da parede vascular e área perivascular, destruição da lâmina elástica interna (vasculite), assim como inflamação do miocárdio e do pericárdio; 6) a disrupção da lâmina elástica interna dos vasos pode condicionar ectasia ou mesmo formação de aneurisma arterial, também apenas em indivíduos com susceptibilidade genética para tal. Após a fase aguda, segue-se a recuperação e regeneração vasculares com processo de aglomeração plaquetária e celular que, a nível vascular, podem levar a trombose e obstrução, com possibilidade de ulterior proliferação da íntima, e de oclusão estenótica.
Segundo os estudos de diversos investigadores, incluindo os da autora (FFP), verifica-se disfunção do endotélio vascular, demonstrada pela redução da produção de óxido nítrico; este fenómeno prolonga-se para além da fase aguda, desconhecendo-se por quanto tempo, ou se existe recuperação completa.
A suspeita de maior predisposição para o desenvolvimento de lesões ateroscleróticas nos doentes que tiveram doença de Kawasaki não foi provada cientificamente, existindo, mesmo, dados que apontam para a ausência de tal associação. Foi, entretanto, demonstrado que em adolescentes e jovens adultos com antecedentes de DK a espessura da íntima-médica das carótidas (parâmetro associado a doença aterosclerótica pré-clínica) é normal.
As ectasias coronárias a longo prazo podem persistir, desenvolver estenoses, calcificar, induzir neoformação de novos aneurismas, ou condicionar oclusão aguda, e até, ruptura coronária.
MANIFESTAÇÕES CLÍNICAS
A febre alta (> 38,4ºC) é característica fundamental (em 100% dos casos), podendo ser remitente e refractária aos antipiréticos. Sem tratamento, em geral dura 1-2 semanas, mas pode manifestar-se durante período curto de 5 dias ou persistir durante 3-4 semanas (ver adiante).
Para além da febre, foram descritos cinco critérios clínicos designados principais, mais frequentes (oscilando em cerca de 50-90% dos casos), e que poderão não ocorrer concomitantemente.
Assim, não existindo exame laboratorial específico para DK, o seu diagnóstico é baseado numa constelação de sinais e sintomas.
Segundo as normas actualizadas (2017) da American Heart Association, o diagnóstico de DK (DK clássica) requer o reconhecimento de febre persistente inexplicada durante um período mínimo de cinco dias*, associada à presença de, pelo menos, quatro dos cinco sinais clínicos seguintes:
- Alterações das extremidades, que evoluem com eritema e edema duro das mãos e dos pés na fase inicial e, posteriormente, descamação em placas (rebatimento de retalhos no sentido distal – proximal dos dedos), uma a três semanas após o início do quadro febril;
- Exantema multiforme envolvendo o tronco e as extremidades, com aspecto variável – urticariforme, simile psoríase, micropustular, morbiliforme ou escarlatiniforme – na primeira semana de doença;
- Hiperémia conjuntival bilateral, indolor, não exsudativa, afectando predominantemente a conjuntiva bulbar e poupando a região límbica;
- Alterações dos lábios e da cavidade oral designadamente: lábios vermelhos e fissurados, língua em framboesa e hiperémia da mucosa orofaríngea;
- Adenomegalias cervicais, mais frequentemente unilaterais, duras e dolorosas e, pelo menos, um gânglio com diâmetro > 1,5 cm.
De acordo com as referidas normas, e valorizando especialmente o achado semiológico de febre:
A DK deve ainda ser considerada se a febre regredir após 7 dias sem tratamento; A DK deve também ser considerada em:- Crianças com < 6 meses, febre prolongada e irritabilidade;
- Lactentes com febre prolongada e meningite asséptica;
- Lactentes ou crianças com febre prolongada associada a linfadenite inexplicada, abcesso ou fleimão retrofaríngeo, ou choque.
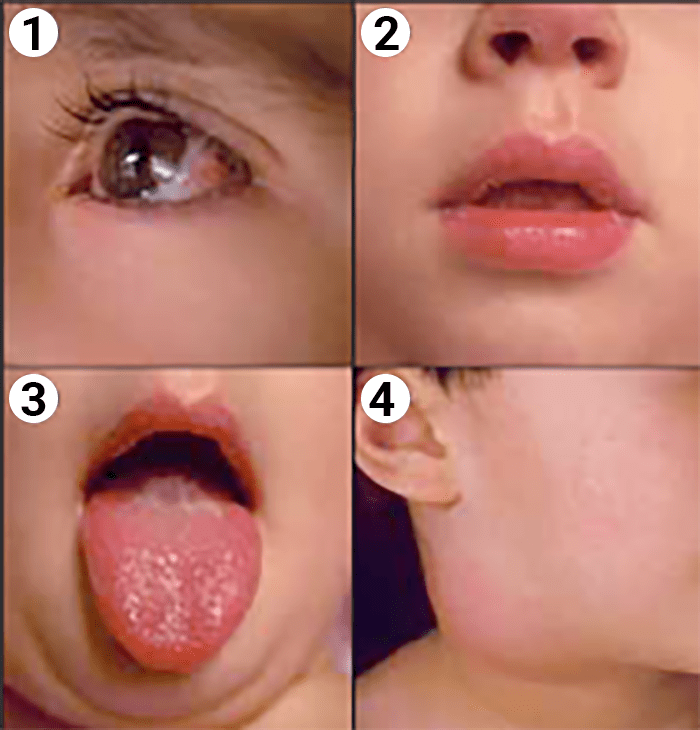
FIGURA 1. Doença de Kawasaki: 1 – hiperémia conjuntival; 2 – lábios vermelhos e fissurados; 3 – língua em framboesa; 4 – adenomegália cervical. (NIHDE)
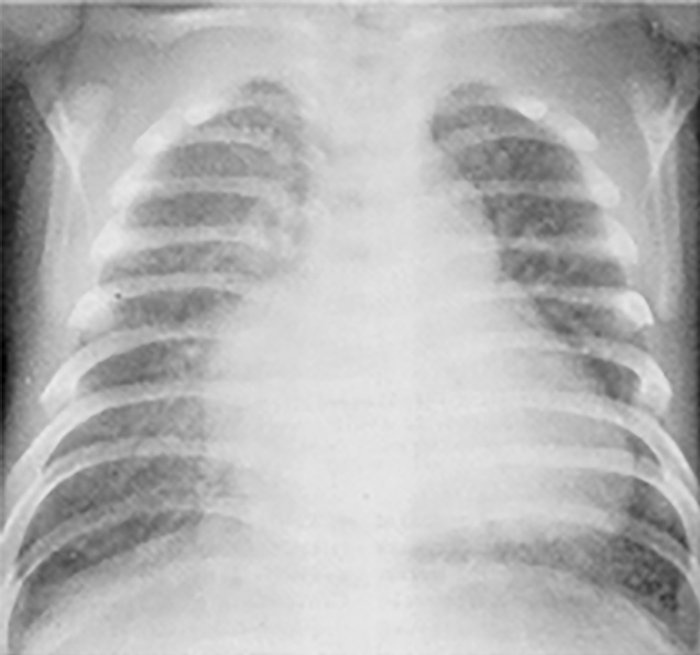
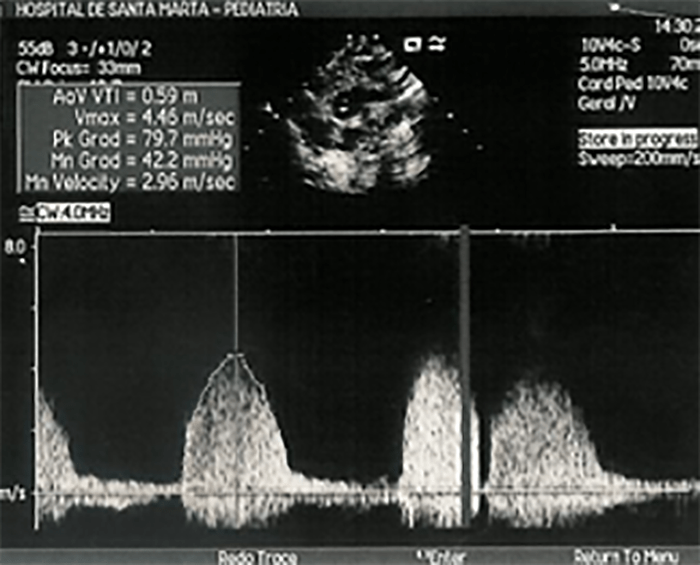
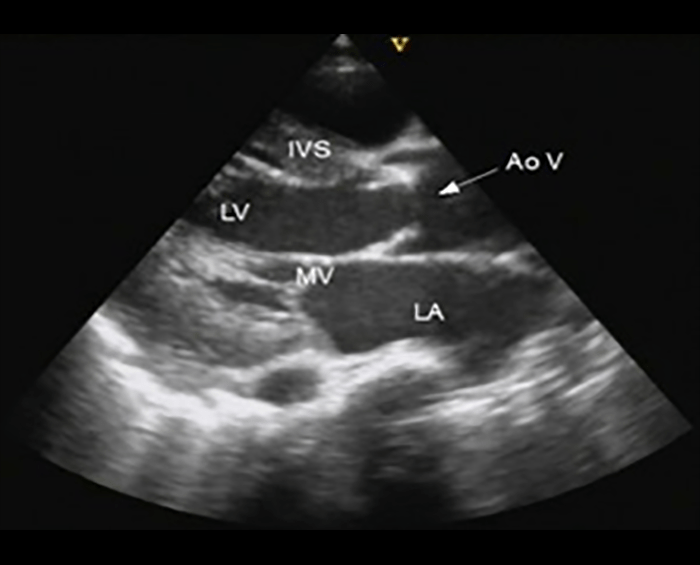
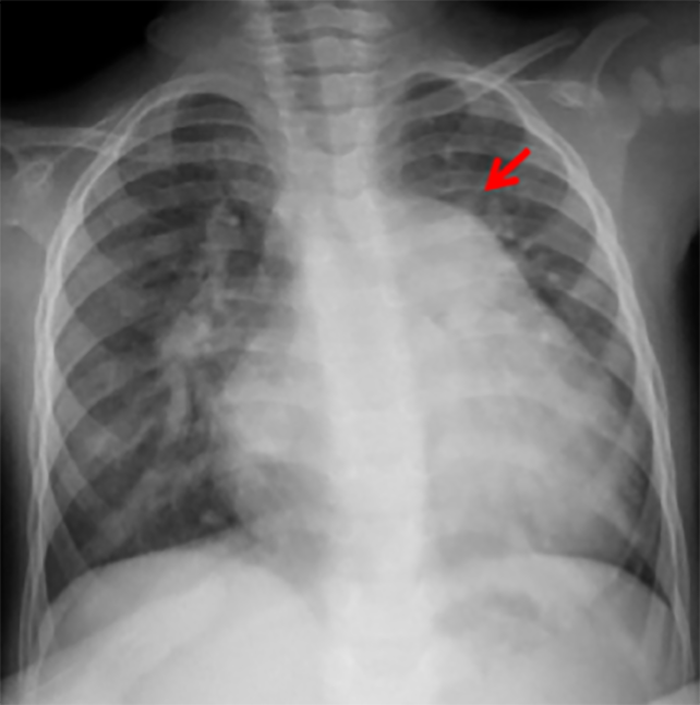
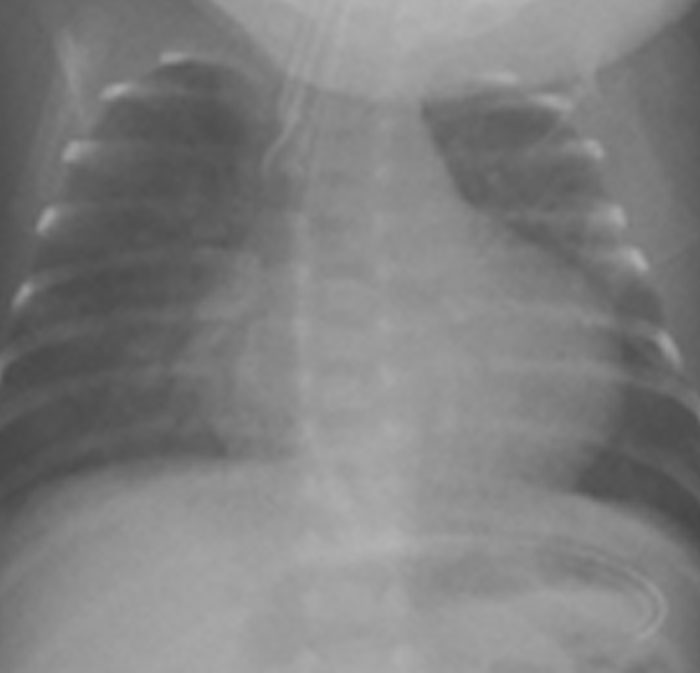
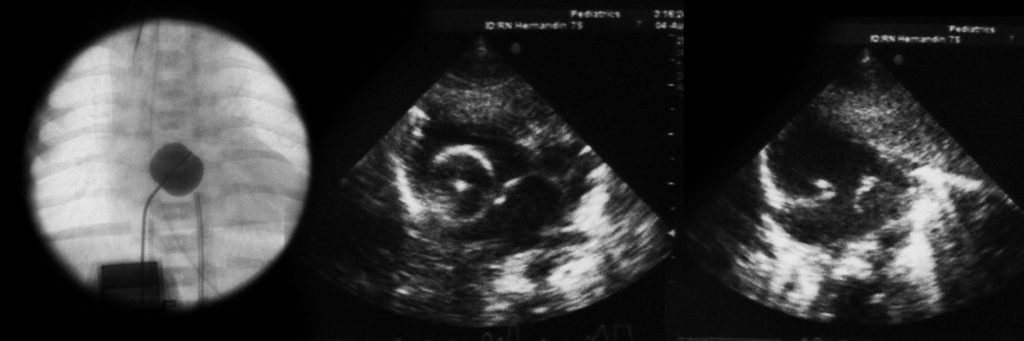
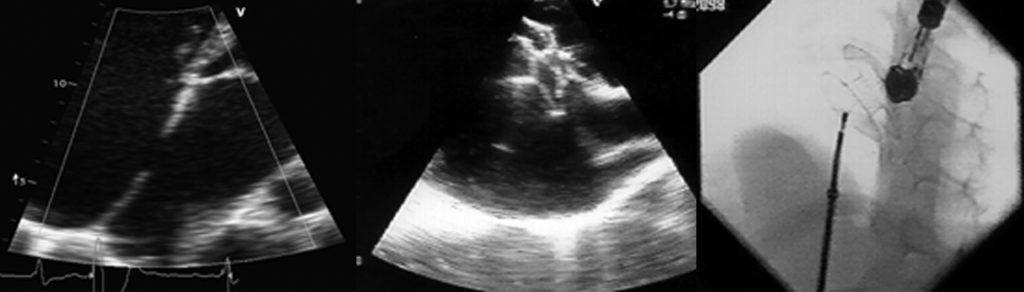
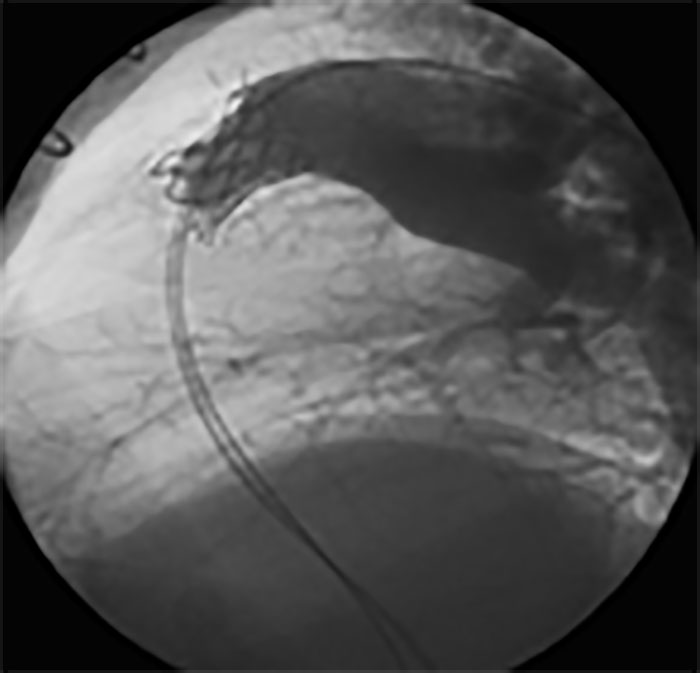
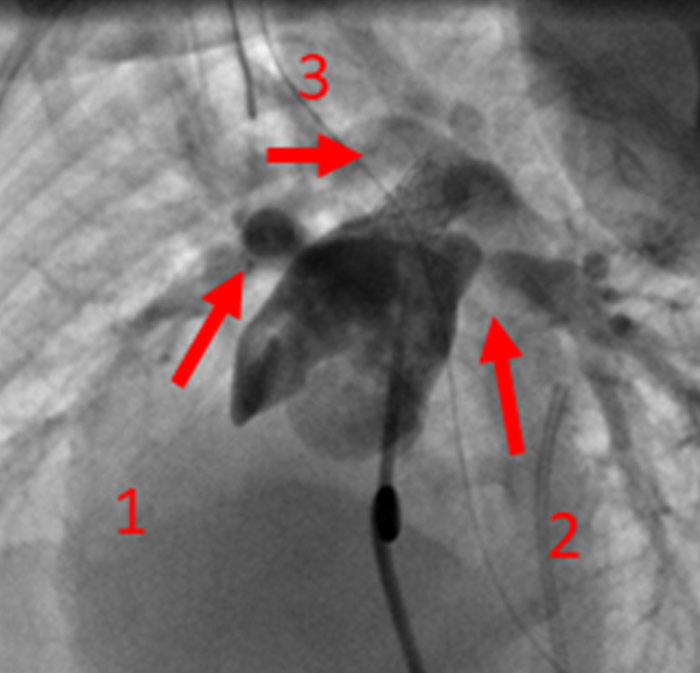
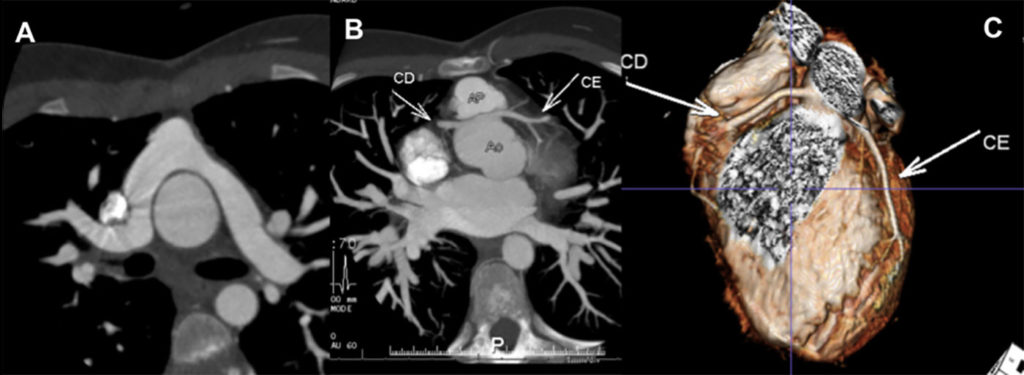
Na valorização das manifestações clínicas, importa considerar, para além dos critérios principais anteriormente descritos, outros achados menos frequentes, associados a DK (~5-40%), tais como: vómitos, diarreia, artralgia, artrite, parotidite, erupção pustular, dor abdominal, paralisia do nervo facial, hepatite, uretrite, leucocitúria estéril, uveíte anterior, iridociclite, hiperémia perianal, hiperémia da cicatriz de BCG, hidropisia vesicular por vezes traduzida por massa palpável no hipocôndrio direito (Figura 2. A-D)
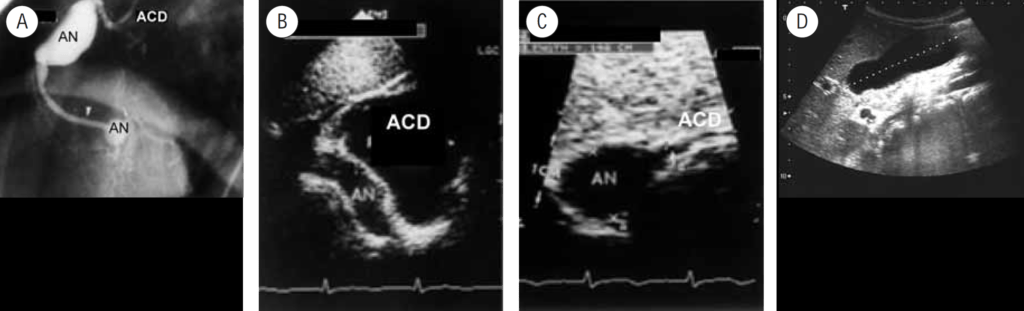
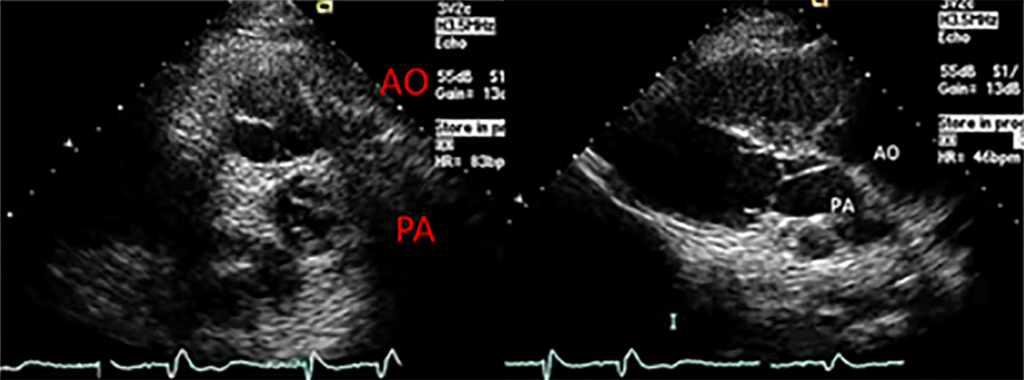
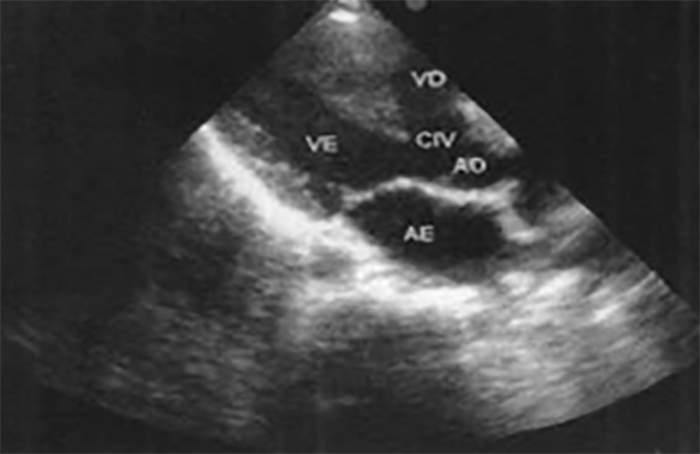
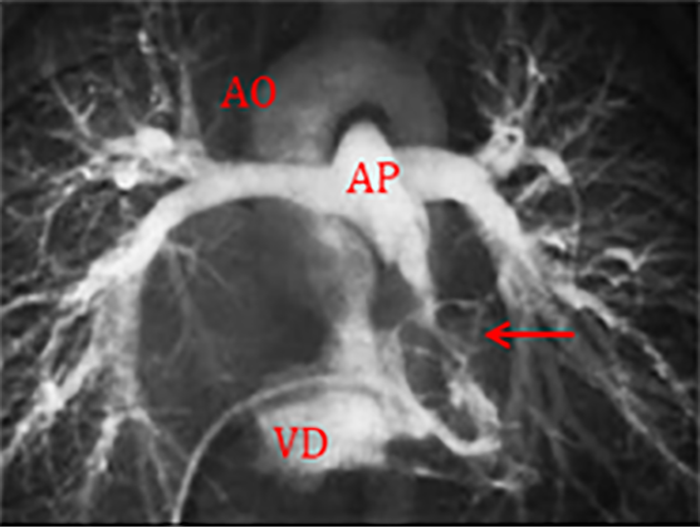
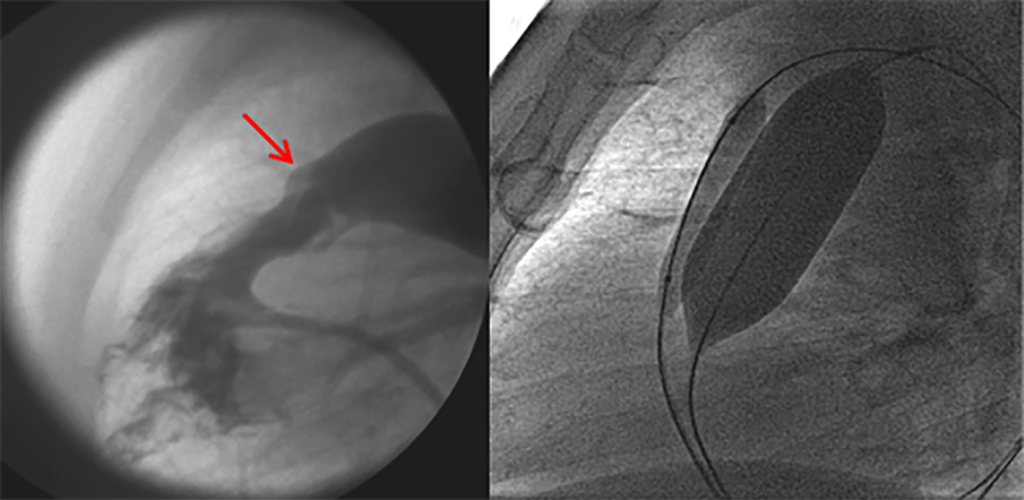
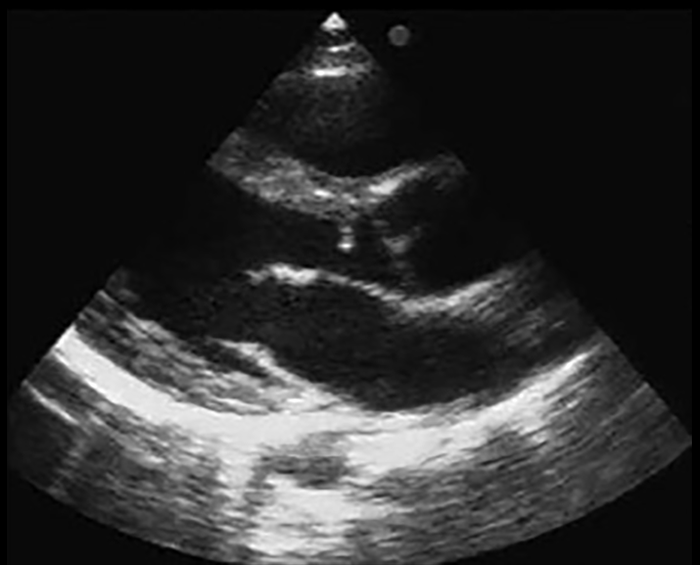
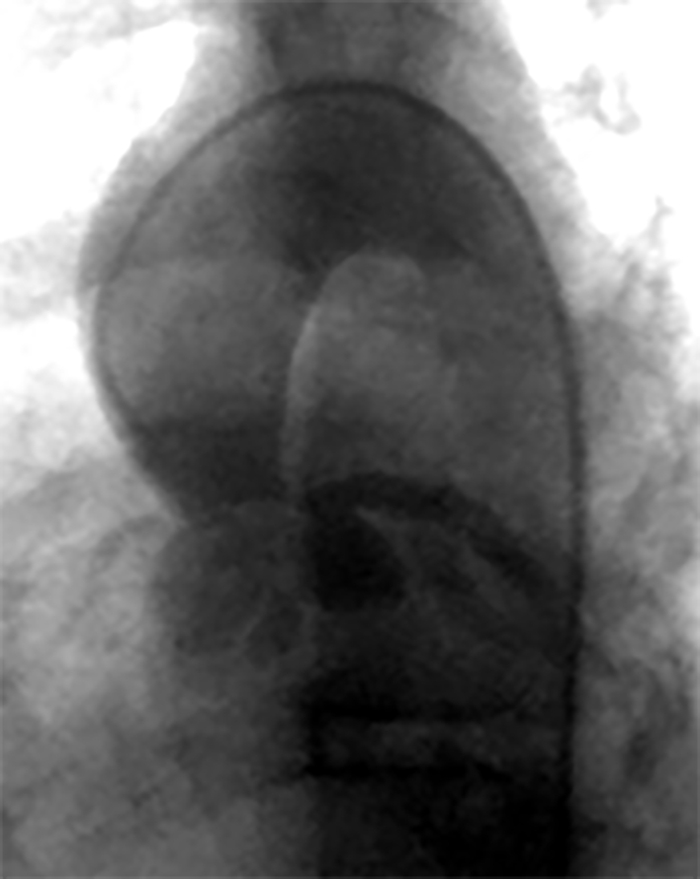
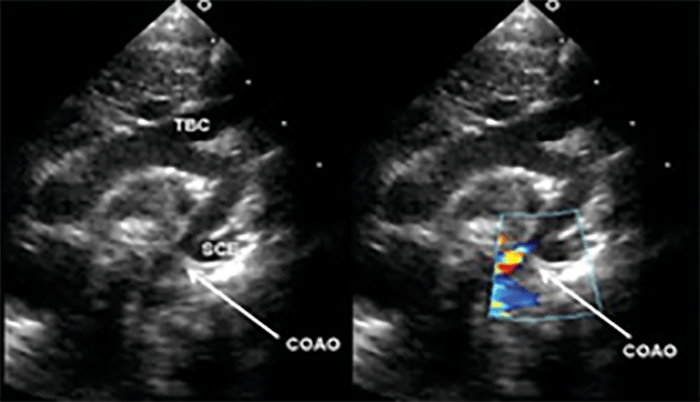
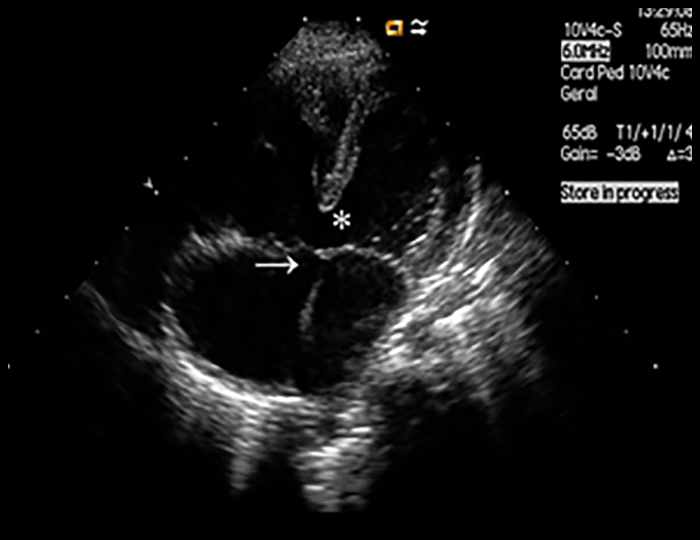
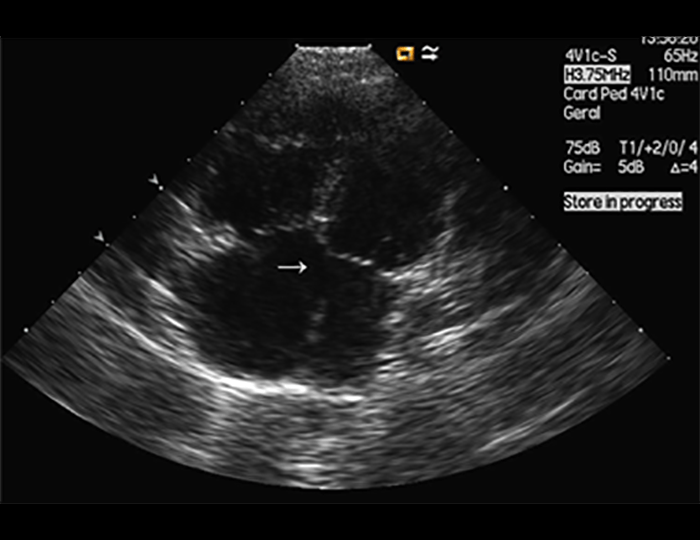
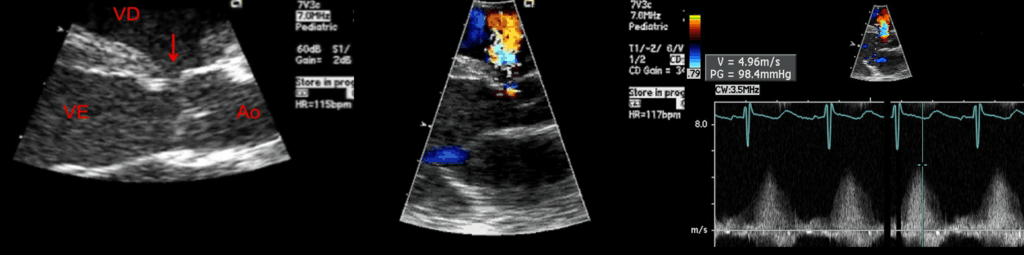
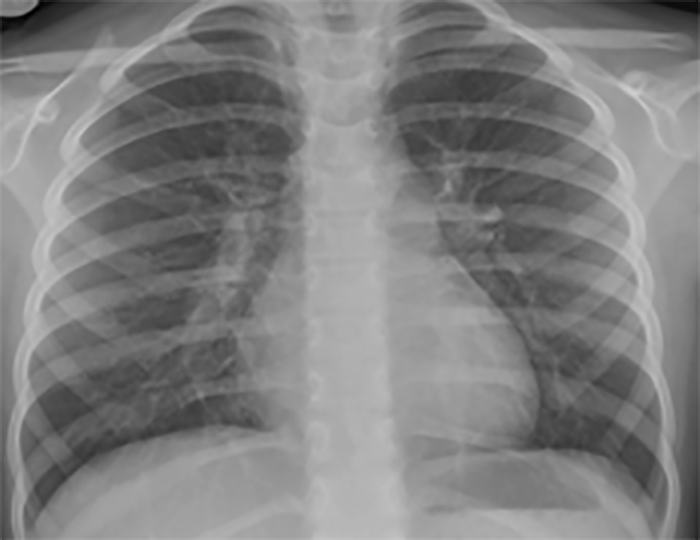
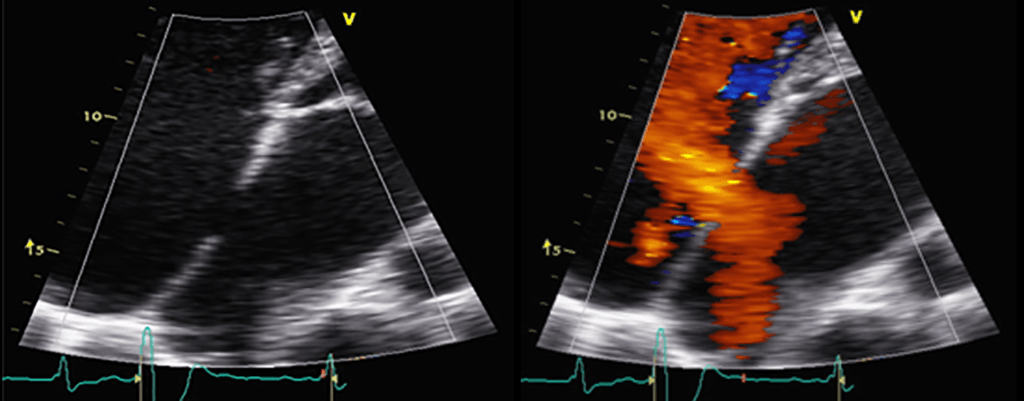
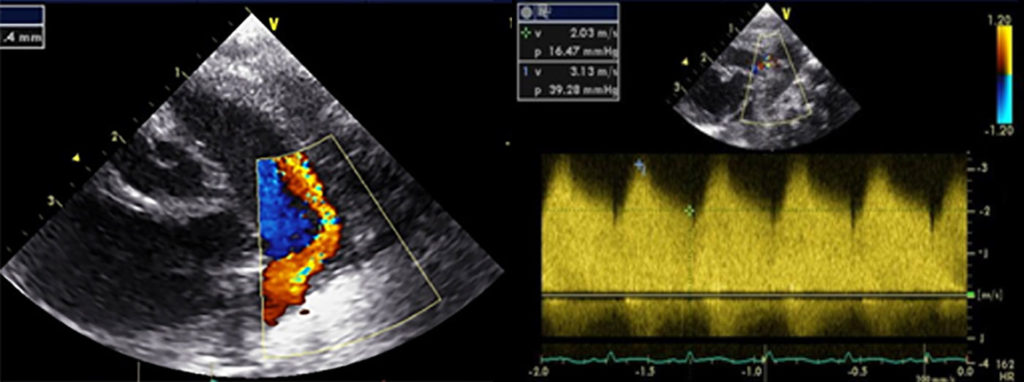
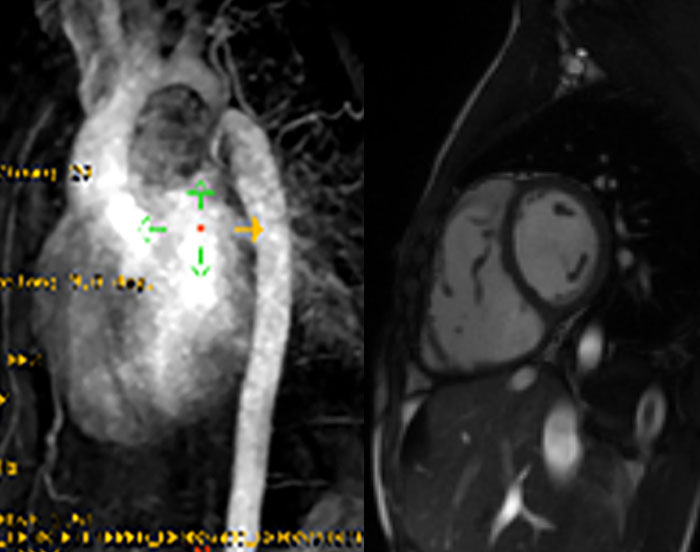
FIGURA 2. DK – Aspectos imagiológicos de aneurisma da artéria coronária direita (ACD): A -arteriografia; B e C – ecografia. D – sinal ecográfico de hydrops da vesícula biliar. (ACD – artéria coronária direita; AN – aneurisma)
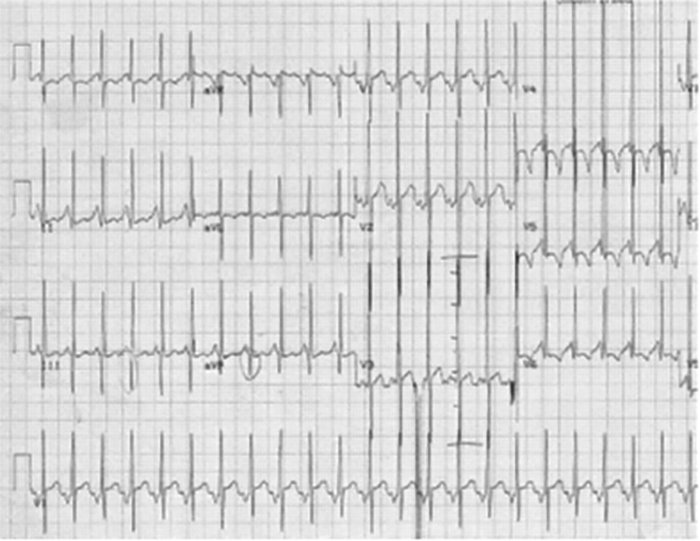
Importa relevar que o envolvimento cardíaco constitui a manifestação mais importante da DK. Arritmias, ritmo de galope ou taquicardia desproporcionada para a febre podem ser uma manifestação de miocardite. Sinais de regurgitação mitral, derrame pericárdico e de choque cardiogénico poderão surgir na fase aguda.
Efectivamente, as lesões cardíacas são as mais preocupantes por constituírem o principal factor de risco de mortalidade e morbilidade, podendo verificar-se, em proporção variável até 25% dos doentes não tratados, a ocorrência de anomalias das artérias coronárias (grandes aneurismas).
Por outro lado, a presença de determinados achados poderá excluir a hipótese de DK: conjuntivite e faringite exsudativas, linfadenopatia generalizada, lesões ulcerosas bucais ligeiras, esplenomegalia e exantema petequial ou vesicular.
INVESTIGAÇÃO LABORATORIAL E IMAGIOLÓGICA – ASPECTOS FUNDAMENTAIS
Apesar de não existir um padrão específico de achados de exames complementares, a conjugação de determinados dados laboratoriais, electrocardiográficos e imagiológicos (ecocardiográficos e radiológicos torácicos) pode sugerir o diagnóstico de DK. (Quadro 1)
QUADRO 1. Critérios de diagnóstico de doença de Kawasaki (DK)
| Nota: Na presença de febre e de alterações cardíacas detectadas pelo ecocardiograma, são aceites menos de 4 dos 5 critérios descritos. (entre parênteses a percentagem média de aparecimento das manifestações clínicas). |
| Febre inexplicada durante ≥ 5 dias (critério essencial), mais 4 das seguintes manifestações: |
| 1. Conjuntivite (hiperémia conjuntival bulbar bilateral, não supurada) (80-90%) |
| 2. Linfadenomegália cervical anterior, aguda, não supurada, > 1,5 cm (50-70%) |
| 3. Exantema polimorfo generalizado, não vesicular, principalmente no tronco (> 90%) |
| 4. Alterações nos lábios e/ou mucosas (80-90%) |
| 5. Alterações das extremidades ou na região perineal (80%) |
| 6. Elevação do péptido natriurético BNP ou da sua porção N-terminal (NT-pro-BNP), alteração que traduz permeabilidade vascular aumentada; trata-se, pois, dum marcador útil na identificação de formas clínicas refractárias à administração de IGIV (ver adiante) |
| 7. Elevação da Troponina I (Tn I), somente numa minoria de doentes com miocardite |
| 8. Elevação da Endotelina I ou IL-17 |
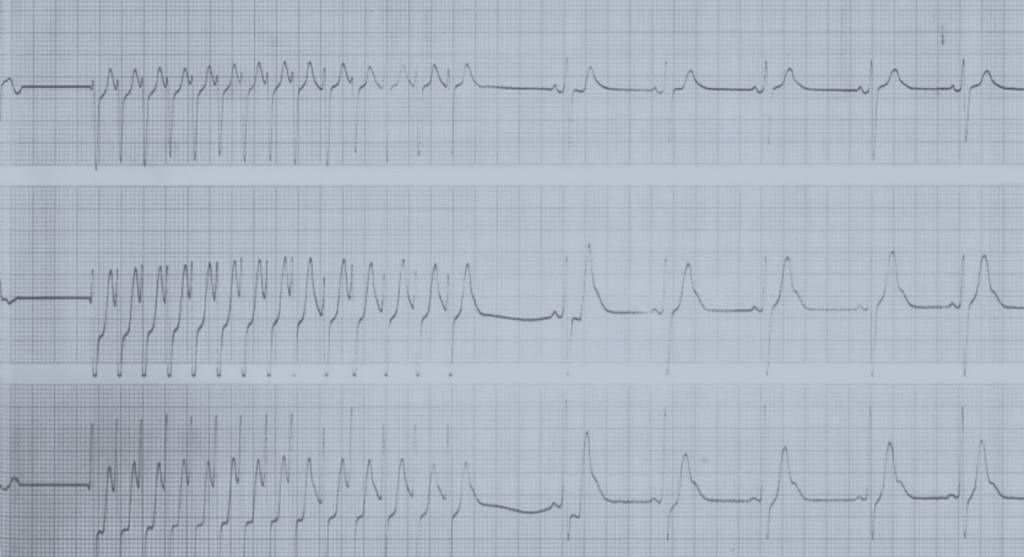
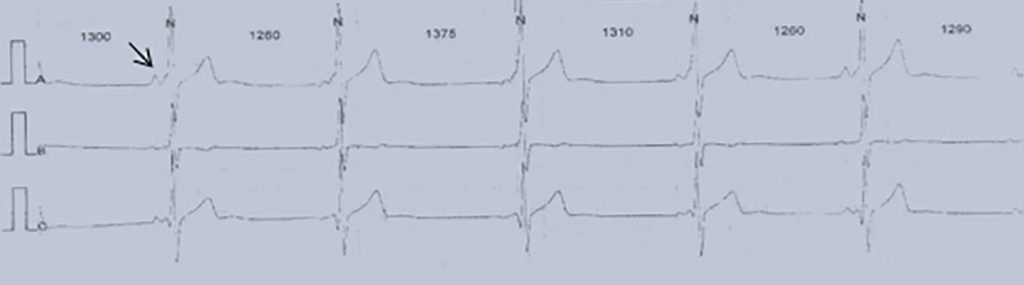
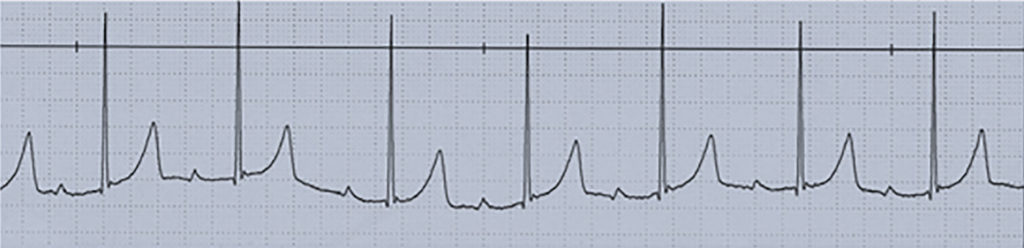
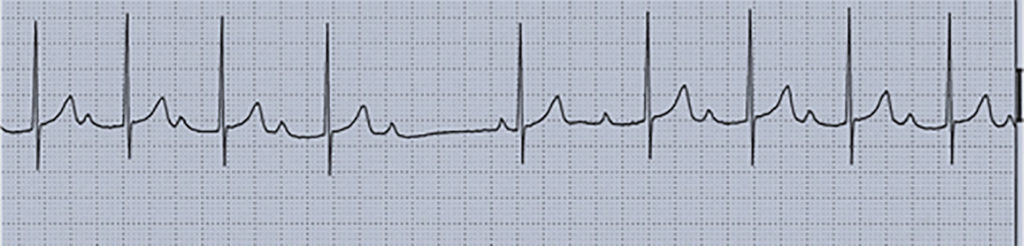
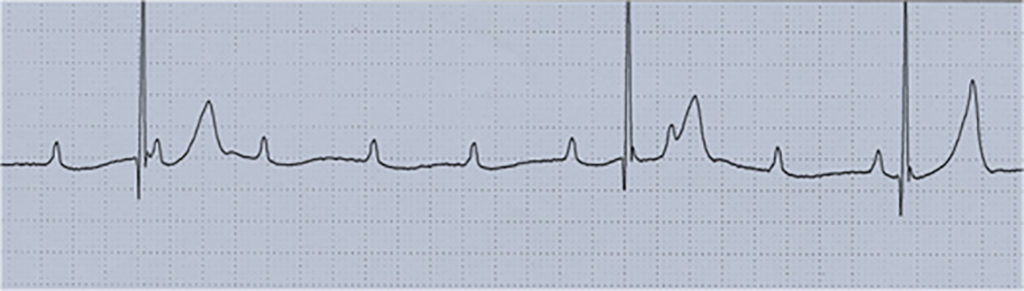
Nota: As alterações electrocardiográficas podem não existir ou ser inespecíficas (redução da voltagem da onda R, depressão do segmento ST com aplanamento ou inversão da onda T, prolongamento do intervalo PQ e/ou do intervalo QT corrigido).
Alguns factores podem estar relacionados com maior risco de aparecimento de aneurismas. O Quadro 2 é elucidativo.
QUADRO 2. Alterações laboratoriais frequentes na DK
| • Anemia normocrómica e normocítica ligeira | • Hipoalbuminémia |
| • Leucocitose com neutrofilia | • Elevação dos triglicéridos e do colesterol-LDL; diminuição do colesterol-HDL |
| • Trombocitose reactiva | • Hiponatrémia |
| • Elevação dos reagentes de fase aguda (VS, PCR e a1-AT) | • Piúria estéril |
| • Elevação da ALT, AST e GGT | • LCR: pleiocitose mononuclear, sem alteração da glicorráquia ou da proteinorráquia |
| • Icterícia obstrutiva (hidropisia vesicular) | • Resultados negativos de exames de culturas e de serologias para agentes infecciosos |
Notas importantes sobre os exames complementares:
|
FORMAS CLÍNICAS
Para além da forma DK Clássica/Típica descrita anteriormente, descrevem-se ainda duas variantes da DK: Incompleta ou Atípica e Resistente
Doença de Kawasaki (DK) Incompleta ou Atípica
Deve ser considerado o diagnóstico de DK incompleta nas seguintes situações:
- Lactentes (especialmente abaixo dos 6 meses), crianças mais velhas e adolescentes com febre inexplicável durante 7 ou mais dias;
- Crianças com febre durante 5 ou mais dias associada a duas ou três das principais características clínicas anteriormente descritas;
- Pode concluir-se que se torna necessário um elevado índice de suspeita para o diagnóstico destes casos, pois o risco de complicações cardíacas aumenta com o atraso do início do tratamento.
Doença de Kawasaki (DK) Resistente
Para a caracterização desta forma clínica, importa considerar aspectos da terapêutica, a abordar adiante).
- Dado que a definição de resistência varia largamente na literatura médica, optou-se por adoptar a definição da AHA: febre persistente ou com recrudescimento durante pelo menos 36 horas após a primeira administração de IGIV;
- Os doentes com DK resistente comportam maior risco de anomalias das coronárias;
- De acordo com a AHA é recomendada uma das seguintes atitudes terapêuticas:
- segunda dose de IGIV
- pulso único de metilprednisolona em alta dose (30 mg/kg) com ou sem seguimento em redução gradual de prednisolona oral
- curso longo de prednisolona oral (durante 2-3 semanas) associado a segunda dose de IGIV
- infliximab
- O regime ideal quanto a dose e duração de corticosteroides no contexto de DK resistente é incerto;
- Outros agentes que têm sido usados na DK resistente incluem: etanercept, anakinra, ciclosporina, e agentes citotóxicos tais como a ciclofosfamida;
- Relativamente à resistência à IGIV, admite-se a existência de genes de maior susceptibilidade para a resistência e para maior risco de lesões coronárias.
EVOLUÇÃO
A DK típica evolui em três fases distintas:
Fase aguda febril (~10 dias): temperatura elevada, irritabilidade, conjuntivite bilateral, exantema, eritema e edema palmar e plantar, orofaringite e queilite. Podem ocorrer disfunção hepática e complicações cardíacas (miocardite e pericardite). O quadro laboratorial clássico, compatível com situação inflamatória aguda, evidencia hipoalbuminémia, trombocitopénia, leucocitose, aumento da velocidade de sedimentação eritrocitária e da PCR (proteína C reactiva).
Fase subaguda (~2-4 semanas): persistência da irritabilidade, anorexia e conjuntivite. A febre tende a desaparecer nesta fase, mas, se persistir, aumenta o risco de complicações cardíacas permanentes. Inicia-se a descamação dos dedos das mãos e dos pés (Figura 3), bem como a formação de aneurismas das artérias coronárias. É a fase de maior risco de morte súbita. Nesta fase, o quadro laboratorial evidencia trombocitose crescente que pode atingir 1.000.000/mm3.
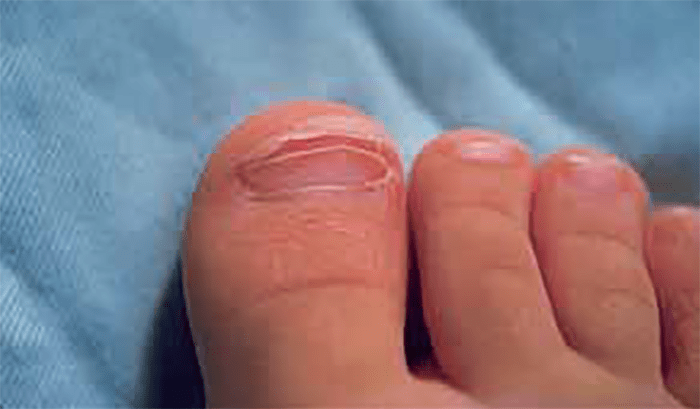
FIGURA 3. Doença de Kawasaki na fase subaguda: descamação periungueal dos dedos dos pés originando o destacamento da epiderme, em lâminas, como que “esfolada”. (NIHDE)
Fase de convalescença ou crónica (~ 6-12 semanas): todos os sinais da doença desaparecem e os marcadores laboratoriais de fase aguda normalizam. Nesta fase, podem persistir, quer os sinais de disfunção endotelial, quer os aneurismas das artérias coronárias. Nos doentes com complicações cardíacas, podem surgir consequências tardias graves, tais como ruptura de aneurismas coronários. A mortalidade global é cerca de 2% nos casos não tratados, e de 0,1% nos tratados. Todavia, a morbilidade e a mortalidade secundárias a complicações cardiovasculares podem ser significativas: nesta perspectiva salienta-se que os aneurismas coronários ocorrem em 20 a 25% dos casos não tratados, o que contrasta com a taxa de 4% nos tratados. Por consequência, o grau de compromisso coronário dita a necessidade de seguimento a longo prazo, sendo a revascularização miocárdica uma prioridade nos casos graves, complicados com estenose ou oclusão coronária (ver adiante).
DIAGNÓSTICO DIFERENCIAL
Como foi referido, a grande dificuldade no diagnóstico de DK reside na ausência de achados patognomónicos que a identifiquem, aliada ao aparecimento sequencial de alterações clínicas e laboratoriais e à possibilidade de quadro clínico incompleto. Por outro lado, a descamação periungueal, que constitui a característica mais facilmente reconhecida, surge apenas numa fase em que as complicações cardíacas já poderão ter ocorrido.
Inúmeras situações podem apresentar etiopatogénese com pontos comuns e evidenciar, por isso, manifestações clínicas sobreponíveis às da DK, nomeadamente de tipo infeccioso e autoimune.
Destacam-se, assim, as situações a ponderar quanto a diagnóstico diferencial:
- Infecção estreptocócica ou estafilocócica (escarlatina, síndroma do choque tóxico, síndroma da pele escaldada);
- Sarampo, rubéola, exantema súbito, infecção por vírus de Epstein Barr, citomegalovírus, enterovírus, influenza A e B e adenovírus; infecção por Mycoplasma pneumoniae; riquetsioses e leptospirose;
- Síndroma de Stevens-Johnson, doença do soro; artrite crónica juvenil, poliarterite nodosa, doença de Reiter (artrite reactiva com manifestações extra-articulares, designadamente conjuntivite).
Muitas destas entidades nosológicas podem ser excluídas clinicamente, uma vez que apenas algumas evidenciam febre que persiste durante mais de 5 dias.
Nos casos de DK verifica-se, por outro lado, grande irritabilidade, o que pode ser explicado pela presença de meningite asséptica. Contudo, esta alteração pode ser observada noutras infecções, nomeadamente no sarampo.
Outro sinal clínico é a presença de eritema e endurecimento no local de inoculação da BCG, provocados pela reacção cruzada entre as proteínas de fase aguda (heat shock proteins) e as células T.
O exantema e as alterações orais e periféricas observados na escarlatina são, por vezes, similares aos da DK, mas naquela não existe conjuntivite. O exantema surge habitualmente no 2º-3º dia de doença, tendo início nas regiões inguinal e axilar, com rápida extensão ao tronco e membros. A descamação ocorre 7 a 10 dias mais tarde. A resposta ao tratamento com antibiótico adequado é habitualmente rápida.
A síndroma do choque tóxico associa-se a situações com mau estado geral, eritema das mãos e pés, exantema difuso e inespecífico da face, descamação do tronco e dos membros, mucosite com compromisso oral, e conjuntivite não exsudativa. Contrariamente, a apresentação inicial da DK não inclui a instabilidade hemodinâmica.
A síndroma de Stevens-Johnson é caracterizada por um eritema multiforme associado a lesões erosivas nas mucosas, nomeadamente, conjuntivas e cavidade oral. O exantema geralmente cede após 10 dias. Se presente, a sobreinfecção bacteriana pode provocar adenomegálias generalizadas.
A doença do soro é uma reacção de hipersensibilidade de tipo III, mediada pela deposição de complexos imunes, com subsequente activação do complemento. Classicamente é provocada por fármacos que, ao actuarem como haptenos, se ligam à albumina plasmática, funcionando como antigénios. Destes, destacam-se: proteínas séricas heterólogas (antitoxinas, gamaglobulina, anticorpos monoclonais), antibióticos (penicilinas, cefalosporinas, sulfonamidas, ciprofloxacina, tetraciclinas, metronidazol), agentes biológicos (estreptoquinase) e outros fármacos (captopril, indometacina, fenilbutazona, procainamida, quinidina, tiouracilo, alopurinol e barbitúricos). Aproximadamente 7 a 10 dias após a administração da substância (coincidindo com o “pico” de complexos imunes circulantes) ocorre febre elevada, mal-estar generalizado e cefaleias. De seguida surge exantema pruriginoso, que tem início no local do inóculo ou, se a via tiver sido oral, com difusão a partir do abdómen. Tal como na DK, é polimorfo (urticariforme, escarlatiniforme, morbiliforme). Dois terços dos doentes apresentam artralgia ou artrite, com predomínio das articulações dos joelhos, tornozelos, ombros e punhos.
A presença de adenomegálias coincide com o início da restante clínica; as cadeias mais afectadas correspondem aos gânglios de drenagem do local de administração do fármaco. Outras alterações clínicas incluem: sinais de compromisso renal (albuminúria, hematúria microscópica, diminuição transitória da depuração da creatinina), edema, sintomas gastrintestinais (náuseas, vómitos, dor abdominal), hepatoesplenomegalia, derrame pericárdico e dificuldade respiratória.
A forma sistémica da artrite idiopática juvenil pode apresentar-se com febre prolongada, sinais sistémicos e artrite.
Para exclusão de algumas das situações descritas, e paralelamente à cuidadosa caracterização clínica, a realização de determinados exames complementares poderá ser equacionada em função de cada caso específico.
TRATAMENTO
Tratamento da doença propriamente dita
Não sendo conhecido nenhum agente etiológico, o tratamento da DK é dirigido para o controlo dos fenómenos inflamatórios, de forma a evitar aparecimento de anomalias das artérias coronárias e as consequentes lesões miocárdicas.
O plano terapêutico consiste na administração de:
1. Imunoglobulina intravenosa (IGIV): (preferencialmente nos primeiros 10 dias de doença), na dose única de 2 g/kg, por via endovenosa, em perfusão de 10-12 horas. Embora o mecanismo exacto de acção seja desconhecido, está comprovado o seu efeito benéfico na rapidez de resolução da fase inflamatória e na prevenção de aneurismas coronários. Em cerca de 15% dos casos verifica-se ausência de resposta/resistência à IGIV (persistência de febre ou recrudescimento do quadro clínico após 36 horas), aspecto que também parece ter predisposição genética. Em tais circunstâncias está indicada a repetição da dose de 2 g/kg.
2. Ácido acetilsalicílico (AAS): em dose anti-inflamatória# (80-100 mg/kg/dia por via oral, em quatro tomas). Esta dose é mantida até ao 3º dia de apirexia, sendo então reduzida para dose antiagregante plaquetária (3-5 mg/kg/dia). Este tratamento pode ser interrompido entre a 6ª e 8ª semanas de evolução nos casos em que não se evidenciem anomalias das artérias coronárias por ecocardiografia. Nos doentes com alterações das artérias coronárias, a antiagregação plaquetária deve manter-se indefinidamente com a finalidade de prevenir trombose coronária.
Os efeitos colaterais do AAS, bem definidos, incluem o aumento das transaminases séricas, hipoacúsia transitória e, raramente, síndroma de Reye. Deve ser interrompido caso se verifique exposição a varicela ou influenza. Em alternativa poderá utilizar-se clopidogrel na dose de 1 mg/kg/dia, até dose máxima de 75 mg/dia.
#De acordo com as normas da AHA-2017, não estando provado que a dose inicial anti-inflamatória de AAS reduza o risco de aneurismas coronários, tal procedimento não é recomendado; por isso, os peritos recomendam apenas AAS em dose antitrombótica. |
3. Outras terapias
3.1 – Têm sido usadas a metilprednisolona IV, ciclofosfamida e plasmaférese em casos de resistência à IGIV, embora sem resultados consistentes.
3.2 – Nos casos em que se verifica resistência à 2ª dose de IGIV e gravidade clínica, nomeadamente com febre alta persistente ou perante ineficácia dos corticóides, utilizam-se inibidores do receptor do TNF-alfa, infliximab e etarnecep. Quanto a estes agentes, estudos recentes não conclusivos demonstraram, contudo, efeito modesto de dilatação das artérias coronárias.
3.3 – Nalguns centros de cardiologia pediátrica utiliza-se baixa dose de uroquinase endovenosa nos casos complicados, verificando-se sinais de trombose coronária.
Tratamento das complicações cardíacas
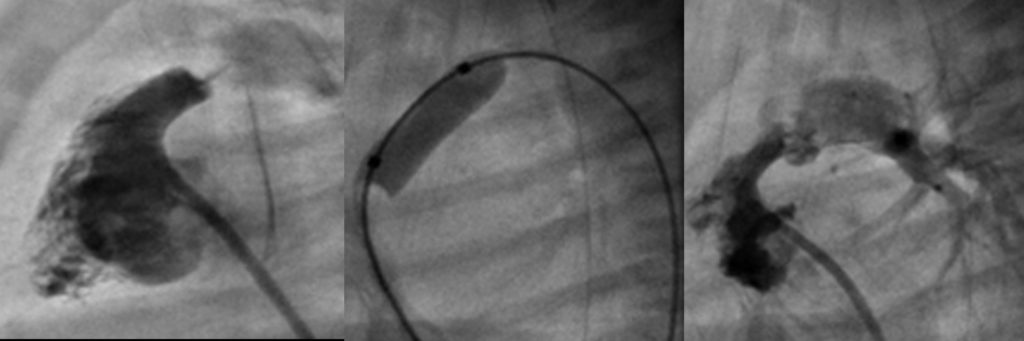
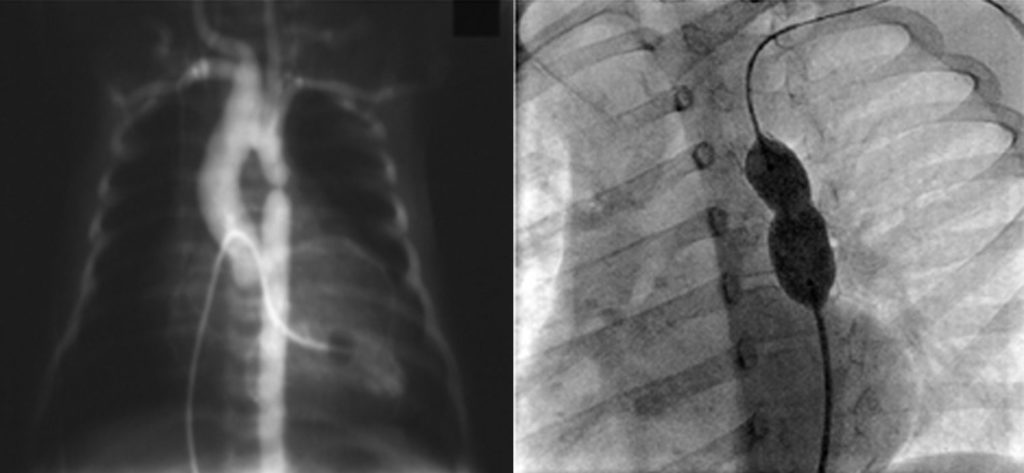
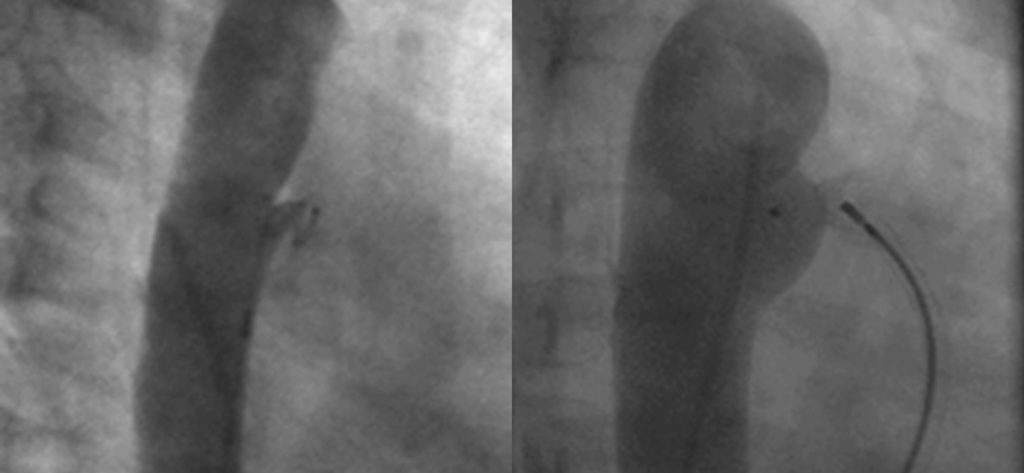
O enfarte agudo do miocárdio é a complicação mais temida e a principal causa de mortalidade. Embora possa ocorrer na fase aguda, é mais habitual na fase subaguda e crónica, quando as lesões coronárias sofrem remodelação. Pode inclusivamente ocorrer mais tardiamente em consequência destas lesões. A experiência da angioplastia coronária por via percutânea é muito limitada em crianças, pelo que nos casos mais graves está indicada cirurgia de revascularização coronária ou transplantação cardíaca.
SEGUIMENTO E PROGNÓSTICO
A actuação a longo prazo deve ser adequada ao grau de compromisso coronário. Existem critérios de estratificação de risco que determinam recomendações relativas a terapêutica antiagregante e hipocoagulante, actividade física e vigilância cardiológica.
Salienta-se que o ecocardiograma deve ser realizado precocemente na fase aguda da doença, e 6 a 8 semanas, após o seu início com o intuito de verificar a eficácia da terapêutica. Na presença de alterações ecocardiográficas, a periodicidade deste exame complementar deverá ser mais estreita. No caso de recorrência (novo episódio com início 3 meses após o inaugural e após normalização da velocidade de sedimentação), o tratamento deve ser semelhante ao inicial.
O paciente deverá ser observado periodicamente para detecção precoce de arritmias, miocardite, regurgitação valvular e insuficiência cardíaca.
A actividade física é limitada pelo próprio doente no período de recuperação, podendo durar algumas semanas. Outras restrições deverão ser impostas apenas nas crianças com risco aumentado de trombose e, particularmente, na presença de aneurismas.
A administração de vacinas vivas atenuadas (como a VASPR e a vacina antivaricela) deve ser adiada, uma vez que os anticorpos adquiridos passivamente através da IGIV persistem até 11 meses, podendo interferir na imunogenicidade. A vacina antivaricela não deverá também ser administrada enquanto durar a terapêutica com salicilatos pelo risco teórico associado de síndroma de Reye. Para diminuição do risco desta síndroma, salienta-se ainda a importância da vacinação antigripe nas crianças submetidas a terapêutica crónica com AAS.
Na presença de um surto de sarampo e caso a criança não esteja imune, a respectiva vacina deve ser administrada e repetida 11 meses mais tarde. O restante calendário vacinal deverá ser cumprido em obediência ao Programa Nacional de Vacinação.
O prognóstico a longo prazo, designadamente quanto à probabilidade de doença aterosclerótica futura, ainda não está bem estabelecido, uma vez que o seguimento destes doentes está actualmente limitado a cerca de 45 anos. Por outro lado, mesmo naqueles em que não se desenvolvem alterações coronárias macroscópicas, nem se verificam alterações ecocardiográficas, são desconhecidas as possíveis consequências da lesão celular endotelial.
Têm sido relatados casos de adultos com enfarte agudo do miocárdio ou outra patologia cardíaca e antecedentes de situação compatível com DK na infância.
É aconselhável proceder a estudo do perfil lipídico cerca de 1 ano após início da sintomatologia de DK, recomendando concomitantemente estilos de vida saudáveis, designadamente alimentação adequada, exercício físico regular e evicção de tabagismo.
AGRADECIMENTOS
O editor e os autores agradecem à Dra. Catarina Gouveia a cedência de uma imagem de ecografia referente a hidropisia vesicular.
BIBLIOGRAFIA
American Academy of Pediatrics. Kawasaki Disease. In Pickering LK, Baker CJ, Long SS, McMillan JA (eds). Red Book: 2006 Report of the Committee of Infectious Diseases. Elk Grove Village, IL: American Academy of Pediatrics, 2006: 412-414
Arane K, Mendelsohn K, Mimouni M, et al. Japanese scoring systems to predict resistance to intravenous immunoglobulin in Kawasaki disease were unreliable for Caucasian Israeli children. Acta Paediatrica 2018; 107: 2179-2184
Baumer JH. Guideline Review. Kawasaki disease: what to do with incomplete cases? Arch Dis Child Educ Pract Ed 2005; 90: ep102 – ep104
Baumer JH, Love S, Gupta A, et al. Salicylate for the treatment of Kawasaki disease in children (Review). The Cochrane Library. London: Wyley, 2009
Breunis WB, Biezeveld MV, Geissler J, Kuipers IM, Lam J, et al. Clinical and Experimental Immunology 2007; 150: 83-90
Burns JC. Kawasaki Disease Update. Indian J Pediatr 2009; 76: 71-76
Burns JC, Mason WH, Hauger SB, et al. Infliximab treatment for refractory Kawasaki syndrome. J Pediatr 2005; 146: 662-67
Burns JC. New perspectives on Kawasaki disease. Arch Dis Child 2019; 104: 616-617
Chen S, Dong Y, Yin Y, et al. Intravenous immunoglobulin plus corticoid to prevent coronary artery abnormalities in Kawasaki disease: a meta-analysis. Heart 2013; 99: 76-82
Durall AL, Phillips JR, Weisse ME, et al. Infantile Kawasaki disease and peripheral gangrene. J Pediatr 2006; 149: 131-136
Fukazawa R e Ogawa S. Long-Term prognosis of patients with Kawasaki disease: at risk for future atherosclerosis? J Nippon Med Sch 2009; 76: 124-133
Gouveia C, Brito MJ, Ferreira GC, Ferreira M, Nunes MAS, Machado MC. Doença de Kawasaki. Casuística do Hospital Fernando da Fonseca. Rev Port Cardiol 2005; 24: 1097-1113
Gray H, Cornish J. Kawasaki disease: a need for earlier diagnosis and treatment.
Arch Dis Child 2019; 104: 615-616
Han SB, Lee S-Y. Antibiotic use in children with Kawasaki disease. World J Pediatr 2018; 14: 621-622
Ito T, Hoshina T, Taku K, et al. Kawasaki disease‐related arthritis with synovial involvement Pediatr Intern 2019; 61: 98-99
Iwashima S, Ishikava T. B-type natriuretic peptide and N-terminal pro-BNP in the acute phase of Kawasaki disease. World J Pediatr 2013; 9: 239-244
Kamath S, Schenck OL, Chamlin SL. Pustular eruption in Kawasaki disease. J Pediatr 2019; 213: 241. https://doi.org/10.1016/j.jpeds.2019.05.018
Kawasaki T, Kosaki F, Okawa S, et al. A new infantile acute febrile mucocutaneous lymphonode syndrome (MLNS) prevailing in Japan. Pediatrics 1974; 54: 271-76
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Kobayashi K, Akishita M, Yua W, Hashimoto M, Ohni M, Toba K. Interrelationship between non-invasive measurements of atherosclerosis: flow-mediated dilation of brachial artery, carotid intima-media thickness and pulse wave velocity. Atherosclerosis 2004; 173: 13-18
Kucera F, Fenton M. Pitfalls in the diagnosis of Kawasaki disease. Paediatr Child Health 2018; 28: 293-294
Lai C-C, Lin W-T, Lin H-C. Parotitis: an initial manifestation of Kawasaki Disease. J Pediatr 2019; 214: 235
Laranjo S, Aidos H, Lourenço A, Rodrigues V, Pinto FF. Ten years of Kawaski in Portugal. National database results. Rev Port Card 2015; 34 (Supl): 67-221
Lima M, Kaku S, Macedo A, Pinto F, Ramos JMS, Magalhães MP, Bento R, Sampayo F. Echocardiographic diagnosis and surgical treatment of giant coronary arterial aneurysms. In Takahashi M and Taubert K (eds). Proceedings of the fourth International Symposium on Kawasaki Disease. American Heart Association. Dallas, 1993: 404-407
Liu Y-C, Lin M-T, Wang J-K, Wu-MH. State-of-the-art acute phase management of Kawasaki disease after 2017 scientific statement from the American Heart Association. Pediatr & Neonatol 2018; 59: 543-552
Makino N, Nakamura Y, Yashiro M, et al. Nationwide epidemiologic survey of Kawasaki disease in Japan. Pediatr Intern 2019; 61: 397-403
Manlhiot C, Niedra E, McCrindle BW. Long-term management of Kawasaki disease: implications for the adult patient. Pediatrics and Neonatology 2012; 54: 12-21
Martins A, Conde M, Brito M, Gouveia C. Arthritis in Kawasaki disease: A poorly recognised manifestation. J Paediatr Child Health 2018; 54: 1371-1374
Masuda H, Kobayashi T, Hachiya A, et al. Infliximab for the treatment of refractory Kawasaki disease: a nationwide survey in Japan. J Pediatr 2018; 195: 115-120
Meena RS, Rohit M, Gupta A, Singh S. Carotid intima-media thickness in children with Kawasaki disease. Rheumatol Int 2014; 34: 1117-1121
Newsburger JW, Sleeper LA, McCrindle BW, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. NEJM 2007; 356: 663-675
Nóbrega S, Santos F, Gouveia C. Síndrome febril e massa palpável no hipocôndrio direito. Actualidades Pediátricas 2008; 11 (2): 17
Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet 2008; 40: 35-42
Phuong LK, Chen KYH, Burgner DP, et al. What paediatricians need to know about the updated 2017 American Heart Association Kawasaki disease guideline. Arch Dis Child 2020; 105: 10-12
Pinto F F, Laranjo S, Paramés F, Freitas I, Carmo M M. Long-term evaluation of endothelial function in Kawasaki Disease patients. Cardiol Young 2013; 23: 517-522
Portman MA, Dahdah NS, Slee A, et al. Etanercept with IVIg for acute Kawasaki disease: a randomized controlled trial. Pediatrics 2019; 143 (6).pii:e20183675. Doi: 10.12542/peds 2018-3675
Rowley AH. Kawasaki Disease: Novel insights into etiology and genetic susceptibility. Annu. Rev Med 2011; 62: 31-39
Santos V, Simões AS, Teixeira A, Abecacis M, Loureiro M, Anjos R. Cirurgia de revascularização coronária após doença de Kawasaki. Rev Port Cardiol 2012; 31: 433-437
Scuccimarri R. Kawasaki disease. Pediatr Clin North Am 2012; 59: 425-445
Senzaki H. Long-Term outcome of Kawasaki Disease. Circulation 2008; 118: 2763-2772
Serizawa H, Lin L, Sato T, et al. Low‐dose i.v. urokinase for coronary thrombosis in Kawasaki disease. Pediatrics International 2019; 61: 302-303
Shiari R. Kawasaki Disease. A review article. Arch Pediatr Infect Dis 2013; 1: 154-159
Son MB, Newburger JW. Kawasaki disease. Pediatr Rev 2013; 34: 151-162
Takahashi M and Taubert K (eds). Proceedings of the fourth International Symposium on Kawasaki Disease. American Heart Association. Dallas, 1993: 404-407
Terreri MT, Clemente G. Developments in large and midsize vasculitis. Rheum Dis Clin North Am 2013; 39: 855-875
Uehara R, Yashiro M, Nakamura Y, Yanagawa H. Clinical features of patients with Kawasaki disease whose parents had the same disease. Arch Pediatr Adolesc Med 2004; 158: 166-169
Yeung RSM. Phenotype and coronary outcome in Kawasaki’s. Lancet 2007; 369: 85-88