Definição
A Algoneurodistrofia Juvenil (ANDJ) é uma síndroma caracterizada por dor extrema habitualmente numa extremidade, em desproporção com o agente causal nocivo, em geral pequeno traumatismo ou imobilização, acompanhada de um ou mais sintomas de disfunção autonómica. Daí, outra designação por que é conhecida – síndroma de amplificação dolorosa.
Foi descrita pela primeira vez na criança em 1971, cerca de um século depois da descrição do adulto. Trata-se duma entidade clínica cujo reconhecimento crescente constitui uma realidade. Para caracterizar tal situação, têm sido utilizadas as seguintes designações (relevando-se em itálico as designações que mais frequentemente surgem na literatura científica): Síndroma de Dor Regional Complexa do tipo I – sem lesão nervosa definida – o habitual na criança e adolescente; e Síndroma de Dor Regional Complexa do tipo II – com lesão nervosa associada) – Complex Regional Pain Syndrome-CRPS; Distrofia Simpática Reflexa – Reflex Sympathetic Distrophy; Distrofia Neurovascular Reflexa; e Atrofia de Sudeck ou Causalgia.
Aspectos epidemiológicos
Sendo a incidência desconhecida, em várias séries pediátricas, a ANDJ é mais comum no género feminino (cerca de 70% dos casos); descrita entre os 5 e os 17 anos e sendo pouco frequente antes dos 7, aponta-se para uma idade média de 13 anos. O tempo médio decorrido entre o início dos sintomas e o diagnóstico varia entre 4 e 12 meses.
Etiopatogénese
Pouco se conhece sobre a etiopatogénese da ANDJ. Parece verificar-se uma activação exacerbada das vias da dor por intermédio dum arco reflexo anómalo transmitido ao córtex cerebral e posteriormente à medula, o que por sua vez determina activação do sistema nervoso simpático, originando manifestações de disautonomia.
A dor e imobilização perpetuam ainda mais este ciclo de nocicepção, com libertação de vários neuropéptidos e mediadores inflamatórios que aumentam a sensibilidade dos axónios lesados. Admite-se igualmente que haja um envolvimento específico do sistema nervoso central a nível dos gânglios da base e lobo parietal. De acordo com um estudo de ressonância magnética funcional, foi identificada a zona do córtex que é activada, permitindo por outro lado demonstrar que, mesmo após a resolução clínica do quadro, se mantêm activadas as vias endógenas da dor.
Factores desencadeantes
Na forma juvenil, o factor desencadeante com acção nociva mais frequente é psicossomático. A história de trauma prévio pode estar presente (cirurgia, fractura, trauma de tecidos moles ou venopunções/injecções), salientando-se que esta circunstância ocorre em menor proporção de casos do que no adulto, é de menor intensidade e pode não estar temporalmente relacionada com o início da dor.
Na criança, sendo de primordial importância o contexto social e familiar, é frequente a associação de ANDJ a antecedentes de abuso sexual, problemas familiares ou na escola, e “bullying”.
Habitualmente verifica-se que os pacientes, com uma “personalidade-tipo”, e com a particularidade de terem assumido cargos de excessiva responsabilidade na família (cuidadores de irmãos mais novos, por exemplo), evidenciam síndromas funcionais (como dor abdominal recorrente, síndroma do cólon irritável, enxaqueca, entre outras), sinais de hipersensibilidade simpática, traduzida designadamente por hipersudorese palmoplantar).
Pode registar-se ainda associação a síndromas de conversão, distúrbios alimentares e tentativas prévias de suicídio. As alterações do sono são muito comuns (insónia inicial e acordares frequentes).
Manifestações clínicas
Do quadro clínico fazem parte a dor intensa e persistente, por vezes descrita como sensação de queimadura, de intensidade crescente e, como se referiu antes, em desproporção com os factores potencialmente nocivos apurados na anamnese e na observação.
Sobre a dor e suas características, importa relevar certos sinais associados:
- Em geral localizada a uma extremidade distal do membro inferior, menos frequente nos membros superiores;
- Recusa de mobilização da região afectada e impotência funcional, num grau aparentemente exagerado;
- Hiperestesia e alodinia frequentes, com intolerância ao simples toque leve e ao contacto da roupa;
- Possível presença de vários sinais de disfunção autonómica, destacando como mais frequentes a alteração da temperatura corporal, o edema das extremidades (não articular e com limites mal definidos), cianose, pele pálida e fria, de aspecto marmoreado e hipersudorese/hiper-hidrose.
No âmbito do adulto foi descrito um escalonamento evolutivo da doença em períodos (os designados Estádios de Steinbroker: I, II e III (Quadro 1). Como particularidade na criança, importa referir que nesta apenas se verificam os estádios 1 e/ou 2 seguinte). Mais adiante estabelece-se com mais pormenor a destrinça entre o quadro clínico no adulto e na idade pediátrica.
QUADRO 1 – Estádios de Steinbroker no contexto da Algoneurodistrofia
| Estádios de Steinbroker |
|
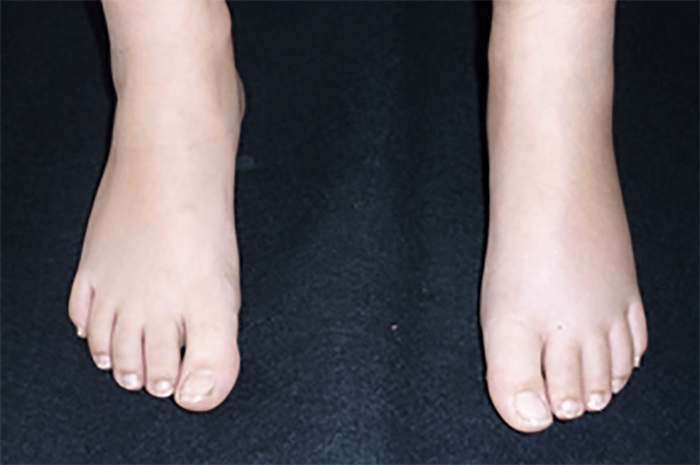
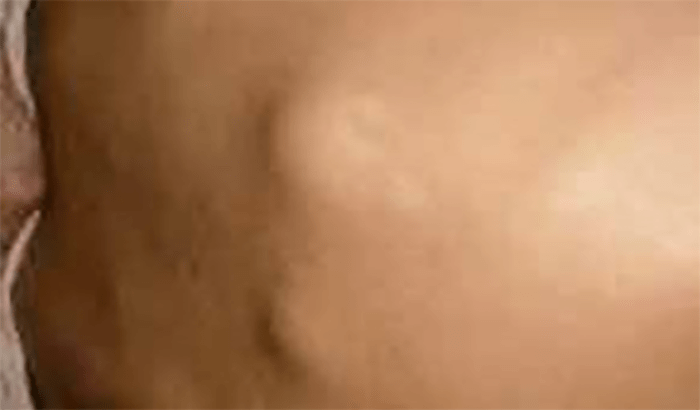
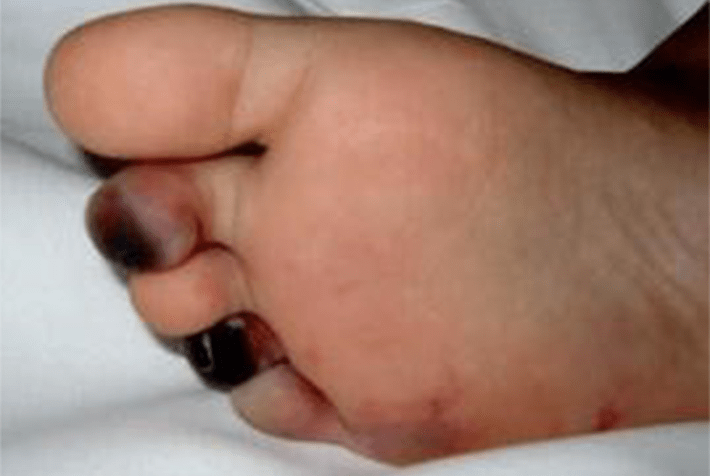
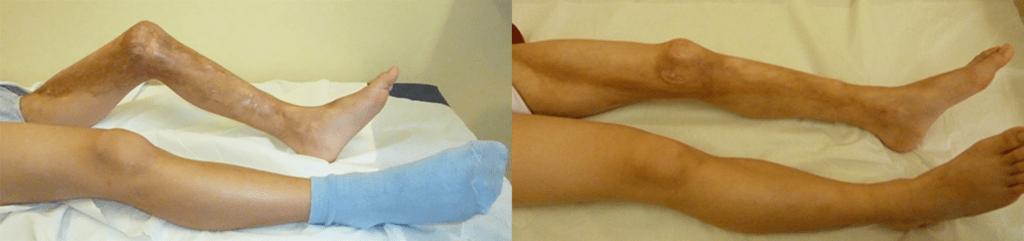
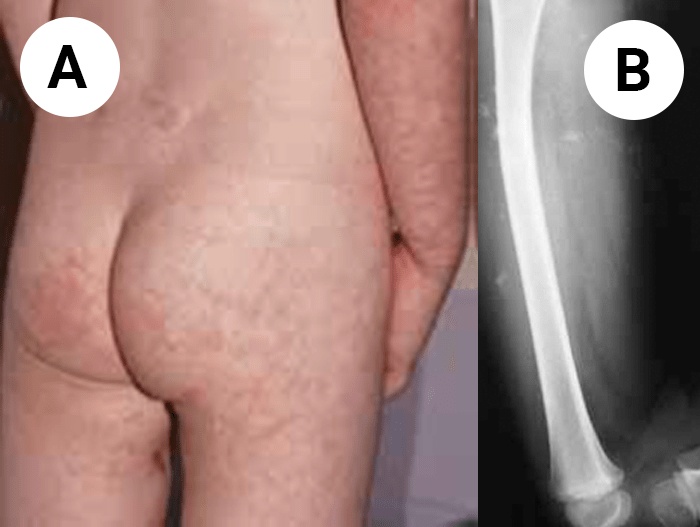
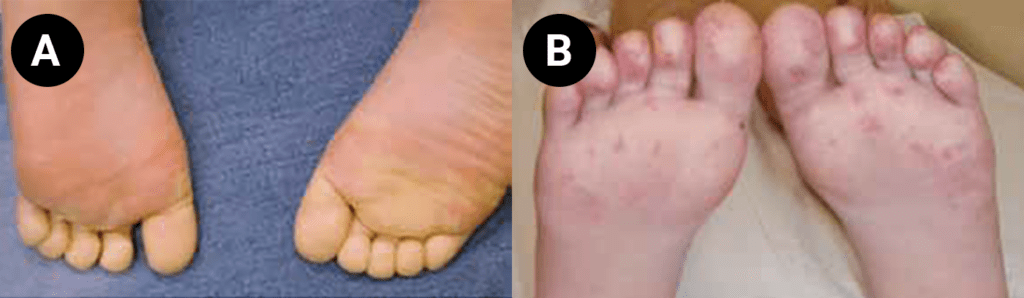
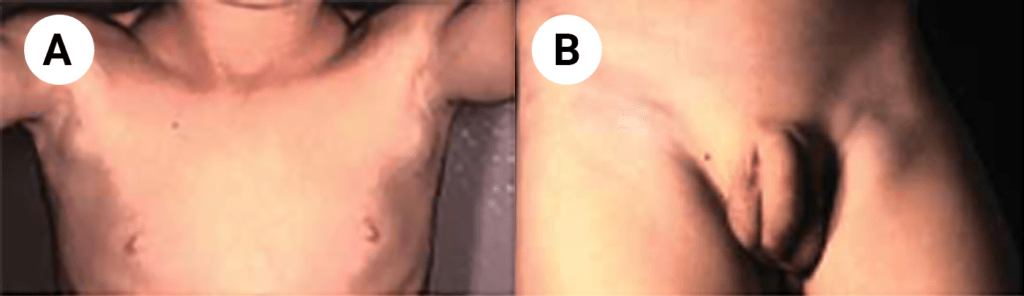
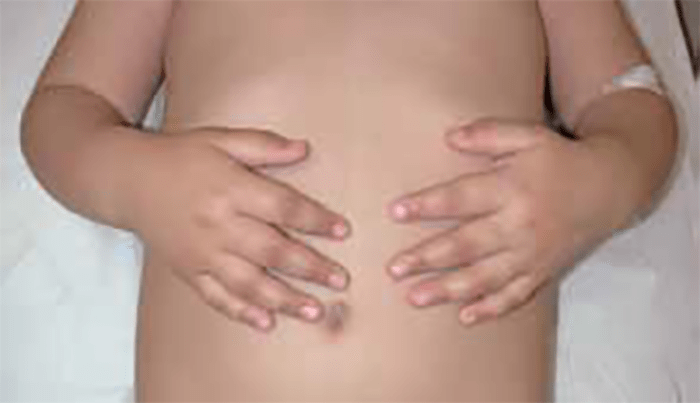
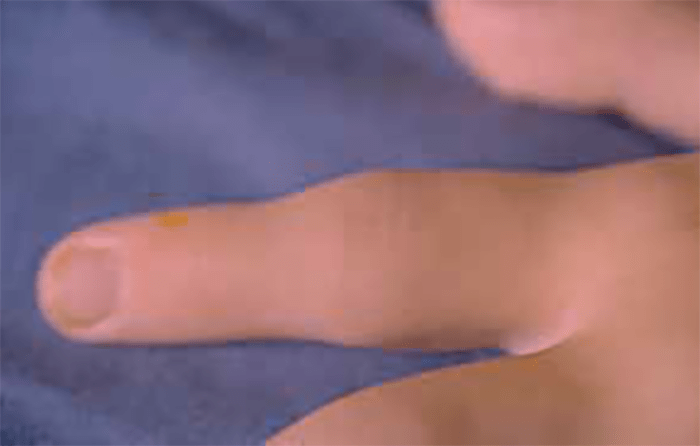
FIGURA 1. Edema de contornos mal definidos, não articular a nível do pé esquerdo em adolescente de 10 anos com AGJ, seguido na Consulta de Reumatologia Infantil, do Adolescente e do Adulto Jovem, Instituto Português de Reumatologia
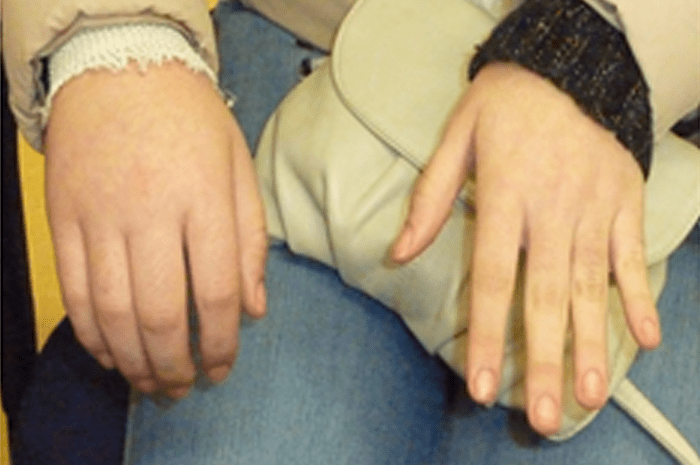
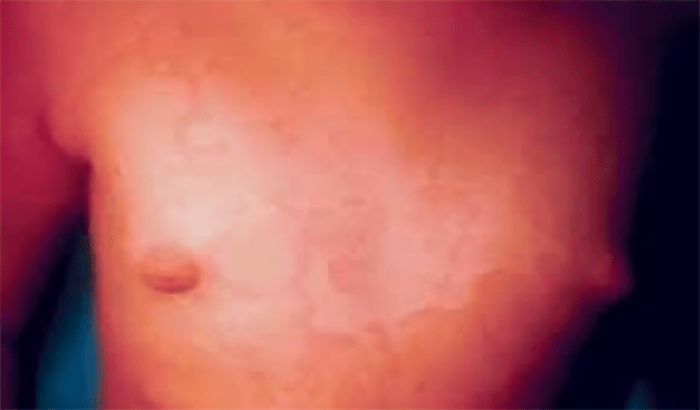
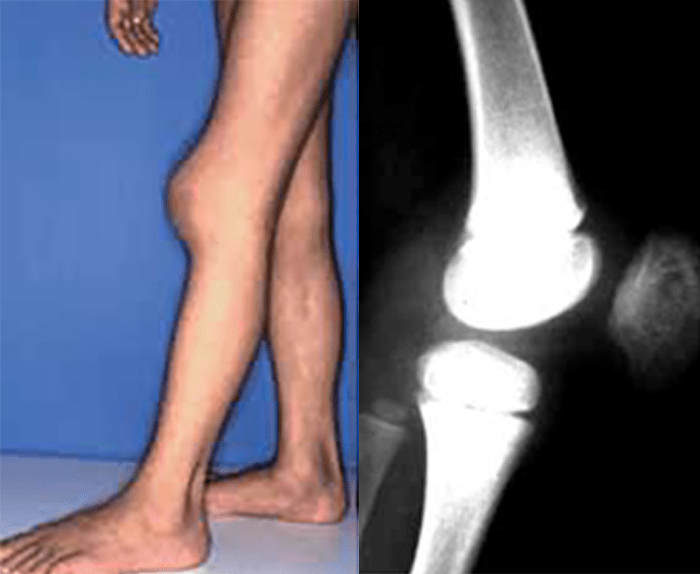
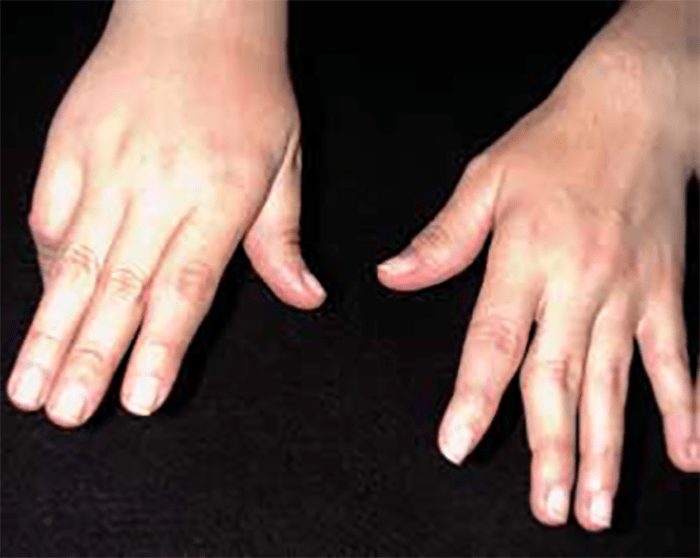
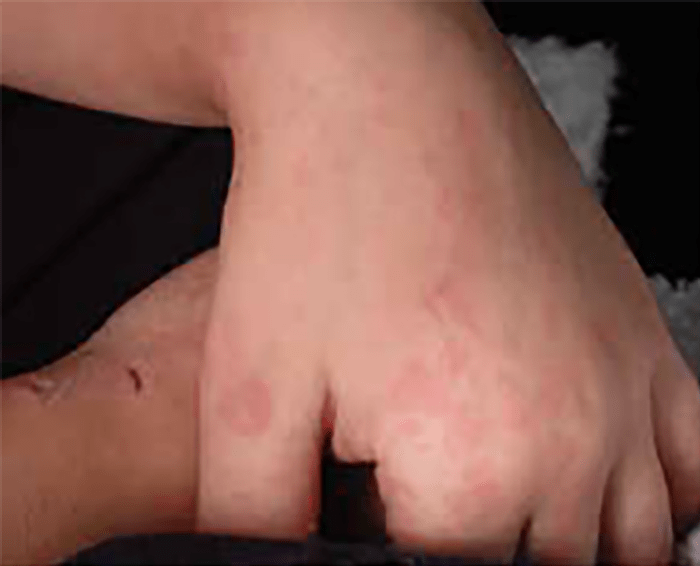
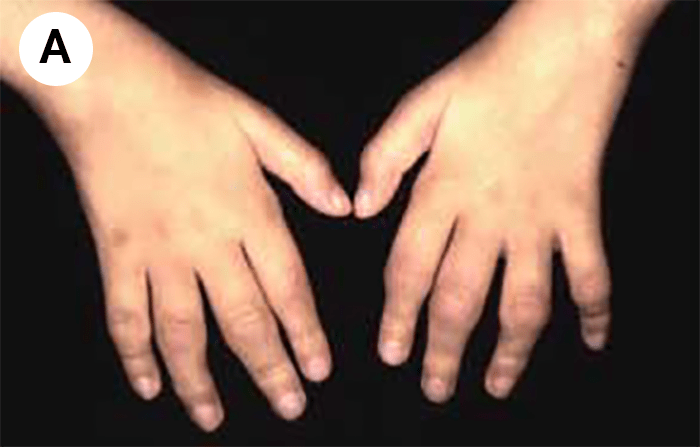
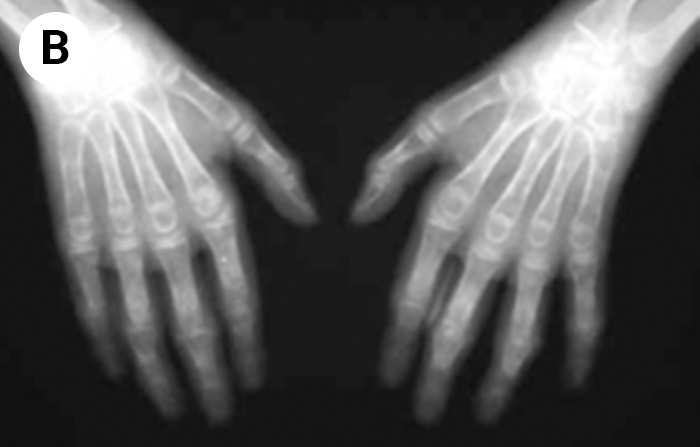
FIGURA 2. Tumefacção difusa da mão direita com impossibilidade de extensão completa dos dedos e uma discreta palidez cutânea em adolescente de 16 anos com AGJ, seguido na Consulta referida na legenda da Figura 1
Exames complementares
Os resultados da avaliação laboratorial, incluindo os parâmetros hemograma completo, velocidade de sedimentação, proteína C reactiva, função renal, hepática e ionograma não evidenciam anomalias, salientando a ausência de elevação dos parâmetros de inflamação.
Verificando-se antecedentes de traumatismo (e supeitando-se de fractura) ou suspeitas de tumor ósseo, está indicado o estudo radiográfico da região afectada. Tal exame, no contexto de AGJ não revela alterações. No adulto, contudo, tal exame poderá evidenciar ocasionalmente sinais de osteoporose mosqueada.
Para exclusão de inflamação articular e outra patologia inflamatória, está indicada a ecografia articular com “Power-Döppler”.
No que respeita a outros exames de imagem, a ressonância magnética pode fornecer achados inespecíficos de edema medular e discreto derrame articular, os quais pouco auxiliam no diagnóstico, sobretudo se houver antecedentes de trauma.
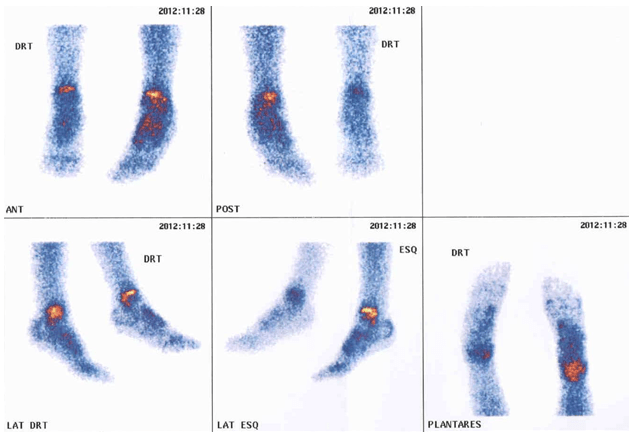
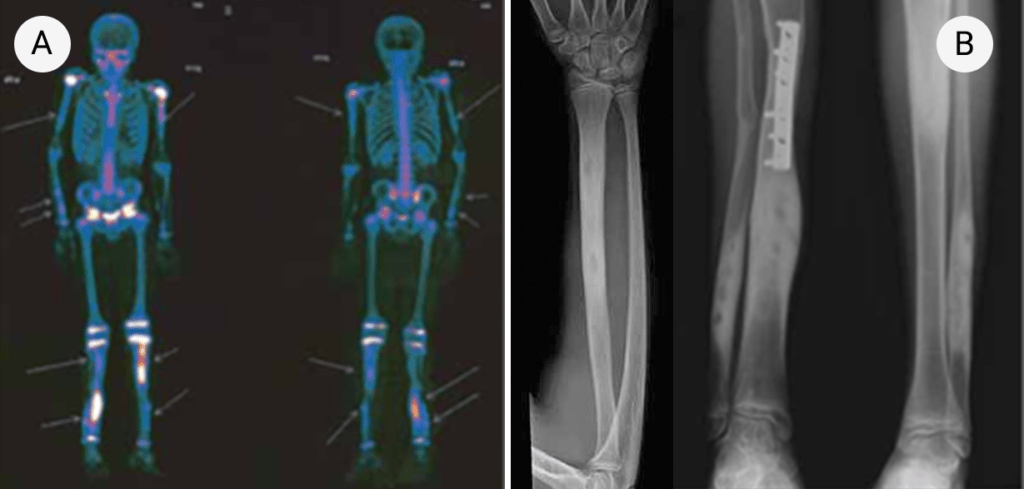
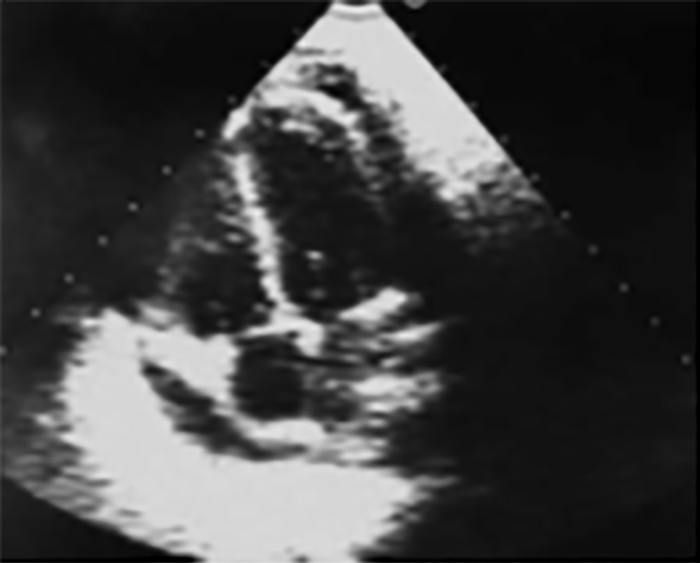
FIGURA 3. Padrão de cintigrafia óssea com 99Tc: hipofixação do radiofármaco ao nível do pé direito num caso de AGJ
O resultado da cintigrafia óssea com 99Tc nos casos de AGJ na idade pediátrica pode evidenciar vários padrões, sendo o mais frequente a hipofixação do radiofármaco na extremidade envolvida, como mostra a Figura 3. Pode também não evidenciar qualquer anomalia ou um padrão de hiperfixação no segmento afectado, tal como é habitualmente observado nos adultos.
Diagnóstico diferencial entre ANDJ na idade pediátrica e no adulto
A forma juvenil tem particularidades próprias em relação à forma do adulto, em termos de factor desencadeante, evolução clínica, resposta ao tratamento, grau de incapacidade e prognóstico funcional, o que leva a colocar a hipótese de que se a ANDJ seja uma entidade totalmente distinta, com base em mecanismos fisiopatológicos próprios. O Quadro 2 resume estas diferenças.
QUADRO 2 – Diagnóstico diferencial entre ANDJ no adulto e na idade pediátrica
| Na Idade Pediátrica | No Adulto |
| Predomínio franco no sexo feminino Idade média: 13 anos | Predomínio ligeiro no sexo feminino Idade média: 46 anos |
| Factor desencadeante psicossomático mais frequente; raramente de tipo “adulto” | Factor desencadeante de etiologia traumática, com ou sem imobilização, ou associado a doença grave prévia |
| Atingimento de membros inferiores | Atingimento de membros superiores |
| Sinais neurológicos e de disautonomia menos frequentes | Sinais de disfunção autonómica mais frequentes e evolução em três estádios |
| Estudo radiográfico normal | Estudo radiográfico: osteoporose mosqueada |
| Cintigrafia óssea com vários padrões (mais comum a hipocaptação na extremidade afectada, excepto nos casos pós-traumáticos) | Cintigrafia óssea com hipercaptação na extremidade afectada |
| Abordagem cognitivo-comportamental por equipa multidisciplinar | Abordagem farmacológica |
| Evolução favorável com medidas de medicina física e reabilitação e apoio psicológico | Fraca resposta a terapêutica conservadora |
Tratamento
Atendendo às particularidades da ANDJ, a abordagem terapêutica deve ser conduzida por uma equipa multidisciplinar que inclua reumatologista pediátrico, pediatra, fisiatra e psicólogo, com o activo envolvimento da criança e dos pais. Os objectivos são restaurar a função, aliviar a dor e quebrar o ciclo da imobilização prolongada.
A terapia ocupacional e física, evidenciando excelentes resultados, deve ser prioritária, e a iniciar logo que seja feito o diagnóstico. Inclui vários tipos de exercícios (aeróbio, hidroterapia, de funcionalidade, etc.) que ajudam na imobilização prolongada e no controlo da dor. Nos estudos em que a terapia física foi utilizada em exclusivo (sem medicação concomitante) verificou-se benefício clínico, designadamente quanto ao restauro da função.
A terapêutica cognitivo-comportamental, fulcral nesta abordagem, tem como objectivos ajudar o doente e a família a tomar consciência da origem dos sintomas e ajudar a criança a aceitar que tem a capacidade de os modificar, tendo o médico um efeito de terapeuta, não antagonizando ou desvalorizando as queixas, mas explicando o processo fisiopatológico e orientando o doente para tomar as suas decisões e vencer a imobilização.
O tratamento farmacológico com analgésicos, antidepressivos ou indutores do sono pode ter alguma relevância, embora os estudos sobre a sua utilização em crianças sejam imprecisos, controversos e sem eficácia documentada.
Os resultados em algumas séries mostram um efeito benéfico com o uso de gabapentina e amitriptilina; contudo, pelos potenciais efeitos, carecem de confirmação com ensaios clínicos controlados.
GLOSSÁRIO
Alodinia > Resposta com dor exagerada por estímulo pouco nocivo ou inofensivo.
Disautonomia > Perturbação funcional do sistema nervoso autónomo ou vegetativo.
Hiperalgesia > Resposta com dor exagerada por estímulo nocivo provocando dor expectável.
Nociceptivo > Diz-se de uma excitação nervosa que provoca sensação dolorosa, ou da reacção provocada por tal excitação (por exemplo, reflexo nociceptivo (reflexo de defesa, tal como a retracção do pé ao ser beliscada a planta)).
Nociceptor > Receptor nervoso sensível aos estímulos produzidos por agentes nocivos (nomeadamente aos estímulos dolorosos).
BIBLIOGRAFIA
Ashwal S, Tomasi L, Neumann M, Schneider S. Reflex sympathetic dystrophy syndrome in children. Pediatr Neurol 1988; 4: 38-42
Barbier O, Allington N, Rombouts JJ. Reflex sympathetic dystrophy in children: review of a clinical series and description of the particularities in children. Acta Orthop Belg 1999; 65: 91-98
Berde CB, Lebel A. Complex regional pain syndromes in children and adolescents. Anesthesiology 2005; 102: 252-257
Bernstein BH, Singsen BH, Kent JT, et al. Reflex neurovascular dystrophy in childhood. J Pediatr 1978; 93: 211-213
Goldsmith DP, Vivino FB, Eichenfield AH, et al. Nucelar imaging and clinical features of childhood reflex neurovacular dystrophy: Comparison with adults. Arthritis Rheumat 1989; 32: 480
Harden RN, Bruel S, Staton H, et al. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8: 326-341
Kachko L, Efrat R, Ben Ami S, et al. Complex regional pain syndromes in children and adolescents. Pediatr Int 2008; 50: 523-526
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Logan DE, Carpino EA, Chiang G, et al. A day-hospital approach to treatment of pediatric complex regional pain syndrome: initial functional outcomes. Clin J Pain 2012; 28: 766-774
Merskey H, Bogduk N. Classification of chronic pain: descriptions of chronic pain syndromes and definitions of pain terms, Seattle: IASP Press, 1994
Sherry DD, Wallace CA, Kelley C, et al. Short- and long-term outcomes of children with complex regional pain syndrome type I treated with exercise therapy. Clin J Pain 1999; 15: 218-221
Sherry DD, Weisman R. Psychologic aspects of childhood reflex neurovascular dystrophy. Pediatrics 1988; 81: 572-574
Silber TJ, Majd M. Reflex sympathetic dystrophy syndrome in children and adolescents. Report of 18 cases and review of the literature. Am J Dis Child 1988; 142: 1325-1328
Tan EC, Zijlstra B, Essink ML, et al. Complex regional pain syndrome type I in children. Acta Paediatr 2008; 97: 875-878
Wilder RT, Berde CB, Wolohan M, et al. Reflex sympathetic dystrophy in children. Clinical characteristics and follow-up of seventy patients. J Bone Joint Surg Am 1992; 74: 910-913
Stanton RP, Malcolm JR, Wesdock KA, Singsen BH. Reflex sympathetic dystrophy in children: an orthopedic perspective. Orthopedics 1993; 16: 773
Wilder RT. Management of pediatric patients with complex regional pain syndrome. Clin J Pain 2006; 22: 443
Laxer RM, Allen RC, Malleson PN, et al. Technetium 99m-methylene diphosphonate bone scans in children with reflex neurovascular dystrophy. J Pediatr 1985; 106: 437
Bialocerkowski AE, Daly A. Is physiotherapy effective for children with complex regional pain syndrome type 1? Clin J Pain 2012; 281: 81‐91