Definição
A glicose é, por excelência, o substrato metabólico dos tecidos, representando o pilar da produção energética celular. Durante a gestação, ela é fornecida por via placentária, pelo que o seu limite sérico inferior ronda os 55 mg/dL. Contudo, nas primeiras horas após o nascimento, no recém-nascido saudável e com peso adequado à idade gestacional, a glicémia pode variar entre 25 mg/dL e 110 mg/dL. Pelas 72 horas de vida, os valores normais em jejum são já sobreponíveis aos do lactente, da criança e do adulto, ou seja, entre 60 mg/dL e 99 mg/dL. Uma vez concluída esta fase de transição, os mecanismos de homeostasia da glicose passam a ser semelhantes ao longo da vida, sendo as únicas diferenças a sua maior taxa de utilização por parte do organismo em crescimento, e um maior risco de sequelas neurocognitivas secundárias à hipoglicémia num sistema nervoso em desenvolvimento.
O intervalo de normoglicémia é mantido por factores que controlam, quer a produção de glicose, quer a sua utilização, pelo que qualquer estado patológico que comprometa este equilíbrio pode desencadear hipoglicémia. As hormonas responsáveis por esta estreita regulação incluem a insulina, o glucagom, a adrenalina, a noradrenalina, o cortisol e a hormona de crescimento.
Apesar de a hipoglicémia ser um fenómeno frequente, em rigor, não é possível definir um valor de corte abaixo do qual esta possa ser estipulada. Na verdade, a hipoglicémia é definida como a concentração plasmática abaixo da qual o indivíduo apresenta sintomas resultantes do fornecimento inadequado deste substrato aos tecidos-alvo, nomeadamente ao sistema nervoso central (SNC). Desta forma, deve ser considerada como um contínuo e os valores plasmáticos devem ser interpretados no contexto clínico e tendo em conta a activação das respostas hormonais de contrarregulação e os respectivos metabólitos intermediários. Classicamente, é caracterizada pela tríade de Whipple: 1. sinais e sintomas consistentes com hipoglicémia; 2. baixa concentração plasmática de glicose; 3. resolução clínica após aumento da glicémia.
As respostas de defesa autonómica e hormonal são desencadeadas quando a glicémia atinge valores entre 69 mg/dL e 55 mg/dL, ocorrendo manifestações major de neuroglicopénia abaixo de 50 mg/dL. Assim, glicémias inferiores a 55 mg/dL constituem uma emergência médica e requerem reversão imediata, sendo comummente aceite um objectivo terapêutico entre 70 mg/dL e 110 mg/dL. No caso de não ser reconhecida e tratada atempadamente, a hipoglicémia pode ter consequências neurológicas graves, particularmente no lactente e na criança pequena. É ainda de extrema importância diagnosticar a etiologia subjacente, de forma a prevenir episódios recorrentes, diminuindo o risco de morbimortalidade associado.
Homeostase
Apesar dos ciclos frequentes de estado pré e pós-prandial, no lactente, na criança e no adulto, os valores de glicémia em jejum normais são mantidos entre 60 mg/dL e 99 mg/dL.
A insulina tem um papel central na regulação da produção e utilização da glicose durante todo este ciclo. A glicémia plasmática começa a aumentar cerca de 15 minutos após a refeição. Este incremento, associado ao estímulo dos eixos neurogénico e êntero-insular (através do peptídeo inibidor gástrico – GIP – e do peptídeo semelhante ao glucagom 1 – GLP1), estimula a produção de insulina pelas células ß pancreáticas. A glicémia atinge um pico 30 a 60 minutos após a ingestão, iniciando a sua descida até um nadir cerca de 4 a 6 horas depois, e seguindo um padrão temporal sobreponível ao da concentração plasmática de insulina.
Após a refeição, as respostas da insulina e do glucagom vão determinar a magnitude da supressão da produção hepática de glicose. Esta produção endógena pode ser reduzida até 50 a 60%, traduzindo-se pela libertação de cerca de menos 25 g de glicose na corrente sanguínea. Por outro lado, durante este período, também a glicogenólise, a gliconeogénese, a lipólise e a cetogénese estão suprimidas. Os principais tecidos responsáveis pela remoção da glicose da corrente sanguínea são o fígado, o SNC, os músculos, o intestino delgado e o tecido adiposo. A concentração de insulina plasmática determina a extensão da utilização da glicose por parte dos tecidos, com a excepção do SNC. Neste, a utilização depende exclusivamente da sua concentração plasmática.
O estado de pós-absorção contempla o intervalo de 4 a 6 horas após a ingestão. Durante este período é atingido um equilíbrio em que a glicémia é mantida num intervalo normal, uma vez que a produção de glicose iguala o seu consumo, estimando-se que este turnover seja de aproximadamente 4 a 6 mg/kg/min. Neste estado, 80% da utilização de glicose é insulino-independente, especialmente por parte do SNC (50% do total), do rim, do intestino e dos eritrócitos. No decurso desta fase, a interacção entre a insulina e as hormonas da contrarregulação mantém estável a concentração de glicose.
A libertação de glicogénio hepático é controlada pelo glucagom, enquanto a insulina limita os efeitos desta hormona, ao prevenir a lipólise e a proteólise. Outras hormonas da contrarregulação, nomeadamente o cortisol e a hormona do crescimento, têm ainda um papel na sensibilidade dos tecidos periféricos ao glucagom e à insulina (Quadro 1).
QUADRO 1 – Resposta fisiológica à diminuição da glicémia
| (Adaptado de Cryer PE, Williams Textbook of Endocrinology 2016) | |||||
| Glicémia (mg/dL) | Resposta | Glicogenólise | Gliconeogénese | Lipólise | Cetogénese |
| 80 a 85 | ↓ Insulina | ↑ | ↑ | ↑ | ↑ |
| 65 a 70 | ↑ Glucagom | ↑ | ↑ | ↑ | ↑ |
| ↑ Adrenalina | ↑ | ↑ | ↑ | ↑ | |
| ↑ Cortisol | ↑ | ↑ | |||
| ↑ Hormona de Crescimento | ↑ | ↑ | |||
| 50 a 55 | Clínica neuroglicopénica | ||||
| < 50 | Diminuição do nível de consciência | ||||
À medida que o período de jejum se prolonga, os tecidos diminuem a utilização da glicose, aumentando a de ácidos gordos livres e de corpos cetónicos. Nesta fase, há uma redução da libertação de glicose hepática, com inibição da glicogenólise e estímulo da gliconeogénese. Este aumento é explicado pela secreção de glucagom e de cortisol, aliado à redução da produção de insulina, os quais promovem a conversão dos depósitos lipídicos em glicerol e ácidos gordos livres, bem como a degradação das proteínas em aminoácidos. Os ácidos gordos são então transportados até ao fígado, local onde sofrem ß-oxidação mitocondrial, ou onde são re-esterificados em triacilgliceróis e fosfolípidos. A ß-oxidação gera acetil-coenzima A, que pode ser transformada em corpos cetónicos (acetoacetato e ß-hidroxibutirato), ou sofrer oxidação no ciclo de Krebs.
Após um período de jejum nocturno, os principais precursores gliconeogénicos são o lactato, o glicerol e a alanina. Na gliconeogénese, a primeira reacção converte o piruvato em oxaloacetato, e este, em fosfoenolpiruvato. O passo limitante de todo o processo consiste na conversão da frutose 1,6-bifosfato em frutose 6-fosfato. Por fim, esta é transformada em glicose livre, que pode ser então utilizada.
Ao contrário do que acontece nos adultos, nas crianças pequenas o glicogénio armazenado é suficiente apenas para as primeiras 8 a 12 horas de jejum, após as quais a gliconeogénese é a responsável pela manutenção de valores glicémicos normais. Este é o principal motivo pelo qual não toleram períodos prolongados sem comerem. Na criança pequena, proporcionalmente, o encéfalo é muito maior que o do adulto, pelo que a taxa de produção de glicose tem necessariamente que ser mais elevada de forma a responder a esta maior necessidade metabólica. Por outro lado, o adulto tem uma melhor capacidade de adaptação neurológica à utilização dos corpos cetónicos. Este é também o motivo pelo qual a criança é mais vulnerável à hipoglicémia e com consequências neurológicas mais graves.
À medida que o período de jejum se torna ainda mais prolongado, as necessidades energéticas musculares e dos restantes tecidos são progressivamente supridas pelos ácidos gordos livres e pelos corpos cetónicos. No fígado, a oxidação dos ácidos gordos gera corpos cetónicos, que são transportados para os tecidos periféricos como substrato energético alternativo. Para além da glicose e dos corpos cetónicos, o encéfalo não utiliza qualquer outro substrato. Assim, à medida que o jejum se prolonga, de forma a satisfazer as suas elevadas necessidades energéticas, estes metabólitos vão substituindo a glicose como combustível celular neuronal predominante.
Manifestações clínicas
Como explicado previamente, quando a glicémia atinge valores abaixo do limite fisiológico do indivíduo, é desencadeada uma resposta hormonal contrarreguladora. Porém, se esta for ineficaz e a glicémia continuar a diminuir, ocorre um estímulo simpático-adrenérgico (Quadro 1). Por este motivo, as primeiras manifestações clínicas são de natureza autonómica, nomeadamente: ansiedade, astenia, palidez, sudorese, tremor, taquicárdia, taquipneia, náuseas e/ou vómitos. Na criança mais velha e no adulto, ocorre concomitantemente uma resposta comportamental no sentido de procura de alimento. É importante salientar ainda que alguns fármacos, como os bloqueadores ß, e algumas substâncias, nomeadamente a cafeína e a teína, podem, respectivamente, diminuir ou amplificar estas respostas.
No caso de agravamento da hipoglicémia (< 50 mg/dL), surgem então sinais e sintomas de neuroglicopénia, designadamente: irritabilidade, hipotonia, cefaleias, confusão, discurso arrastado, alterações cognitivas e eventualmente, convulsão, coma ou mesmo morte. Nesta fase, os corpos cetónicos constituem o principal substrato energético neuronal, cruzando a barreira hematoencefálica através do transportador de monocarboxilatos 1 (MCT1). Este encontra-se sobre-expresso em indivíduos com cetonémia crónica, motivo pelo qual o jejum repetido e a dieta cetogénica se associam a menor intensidade de sintomas e sinais de neuroglicopénia.
O limiar de resposta neuroendócrina à hipoglicémia varia, não só com a idade e o sexo, mas também com o exercício, o sono e o estado nutricional. Contudo, as diferenças mais significativas decorrem da existência de episódios prévios de hipo ou hiperglicémia. De facto, hipoglicémias prolongadas ou recorrentes podem diminuir de tal forma o limiar de activação da resposta autonómica, que apenas a clínica neuroglicopénica se encontra presente perante uma situação grave, condição designada por insuficiência autonómica associada à hipoglicémia (HAAF). Esta situação preocupante é mais frequente, não só na diabetes insulinodependente, mas também nos doentes com hiperinsulinismo. No extremo oposto, na hiperglicémia crónica o limiar para a resposta contrarreguladora à hipoglicémia é mais elevado.
Diagnóstico diferencial
A hipoglicémia pode dever-se à diminuição da ingestão de glicose (ex. gastrenterite, anorexia), ao aumento da sua utilização (ex. sépsis, hipotermia, exercício intenso, hiperinsulinismo), à diminuição da sua produção endógena (ex.: erros inatos do metabolismo, insuficiência hepática, insuficiência adrenal, défice de hormona do crescimento), ou ser secundária a intoxicação ou a fármacos (ex.: álcool, sulfonilureias, insulina, bloqueadores-ß) (Quadro 2).
QUADRO 2 – Diagnóstico diferencial de hipoglicémia na criança não diabética
| Hipoglicémia Cetótica Idiopática |
|
Hipoglicémia cetótica idiopática
A hipoglicémia cetótica idiopática é um diagnóstico de exclusão, constituindo a causa mais frequente de hipoglicémia no não diabético. Pensa-se que em tal situação as crianças representem a cauda inferior da distribuição gaussiana da tolerância ao jejum, podendo ter polimorfismos ou deficiências parciais das enzimas envolvidas na homeostasia da glicose. A produção hepática de glicose está diminuída, possivelmente pela diminuição do abastecimento de alanina, um dos principais precursores da gliconeogénese.
Tipicamente, ocorre entre os 18 meses e os 7 anos, sendo precipitada por um jejum prolongado no contexto de um episódio agudo intercorrente, por exemplo gastrenterite. Apesar de algumas crianças terem episódios de hipoglicémia suficientemente graves para desencadearem convulsões, nestes casos as sequelas neurológicas são muito raras e o neurodesenvolvimento habitualmente é normal. A hipoglicémia pode ser evitada por refeições regulares, assegurando que não ocorrem períodos de jejum superiores a 12 horas. Com o passar do tempo, verifica-se resolução espontânea, sendo rara após a puberdade.
Causas endócrinas
Hipoglicémia hiperinsulinémica
No decurso de um episódio de hipoglicémia, uma das primeiras respostas do organismo é a suspensão da produção de insulina, pelo que a sua detecção no plasma, mesmo que em pequena quantidade, é considerada anormal.
O hiperinsulinismo congénito é a causa mais frequente de hiperinsulinismo. A sua apresentação ocorre no período neonatal ou nos primeiros meses de vida, durante uma intercorrência que impõe um aumento da necessidade da glicose a que, por desregulação da produção de insulina, o organismo não consegue responder. A incidência global é de cerca de 1:30.000 a 1:50.000 nados-vivos, aumentando para aproximadamente 1:2.500 nados-vivos na Península Arábica. Na sua origem estão identificadas cinco mutações: no receptor 1 das sulfonilureias (SUR-1), no Kir6.2, na glucocinase, na desidrogenase do glutamato (GDH) e na desidrogenase da 3-hidroxiacil-CoA de cadeia curta (SCHAD). Estas mutações traduzem-se por fenótipos muito variáveis: enquanto uns indivíduos apresentam hipoglicémia ligeira durante o jejum e respondendo bem à medicação, outros apresentam hipoglicémia grave e refractária à terapêutica farmacológica.
Nas células ß pancreáticas, o SUR-1 e o Kir6.2 são essenciais para a actividade dos canais de potássio sensíveis ao ATP (KATP), sendo a perda da sua função a causa mais frequente de hiperinsulinismo congénito. Na sua origem estão mutações dos genes ABCC8 e KCNJ11 presentes no cromossoma 11p15. Geralmente são herdadas de forma autossómica recessiva, com aumento pancreático difuso das células ß. Esta é a forma mais grave da doença, que habitualmente não responde a terapêutica médica, necessitando de pancreatectomia. Estão ainda descritos casos esporádicos provocados por mutação homozigótica paterna, com perda dos alelos maternos. Nestas situações, a tomografia de emissão de positrões (PET) com 18F-fluro-L-di-hidroxifenilalanina (18F-DOPA) revela frequentemente uma lesão focal solitária.
Na mutação da SCHAD (também autossómica recessiva), a hipoglicémia hiperinsulinémica está associada a defeitos da oxidação dos ácidos gordos, pelo que estes doentes apresentam aumento dos níveis de 3-hidroxibutiril-carnitina plasmática e de 3-hidroxiglutarato urinário. A causa exacta desta desregulação ainda não está bem esclarecida, sendo a sua apresentação clínica variável e com boa resposta ao diazóxido.
Outras mutações mais frequentemente associadas a hiperglicémia (nomeadamente a Maturity Onset Diabetes of the Young – MODY) também podem provocar hiperinsulinismo transitório, como é o caso dos factores hepatocitários nucleares 4-alfa (HNF4α) e 1-alfa (HNF1α). Por outro lado, mutações activadoras da desidrogenase do glutamato provocam não só aumento da produção de insulina, mas também hiperamoniémia, cuja apresentação é mais tardia e com clínica ligeira.
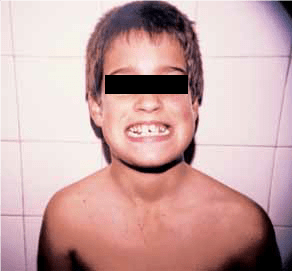
Necessidades de glicose intravenosas superiores a 8 mg/kg/minuto são muito sugestivas de hiperinsulinismo, podendo mesmo ser necessárias perfusões superiores a 20 mg/kg/min. Concomitantemente, deverá tirar-se partido da alimentação entérica, rica em hidratos de carbono. Uma vez estabilizada a glicémia, o tratamento farmacológico de primeira linha é o diazóxido oral (5 mg/kg/dia, em 3 doses; se necessário, com incrementos de 5 mg/kg/dia a cada 4 dias, até um máximo de 20 mg/kg/dia) em combinação com hidroclorotiazida oral (7-10 mg/kg/dia, em 2 doses). Doses de diazóxido mais elevadas, além da demonstração de não eficácia, provocam efeitos colaterais significativos, nomeadamente hipertensão arterial e hipertricose. Se este regime não for suficiente para manter a glicémia acima de 70 mg/dL, será necessário iniciar tratamento com octreótido (5 µg/Kg/dia em perfusão sc contínua; se necessário, com incrementos de 5 µg/Kg/dia a cada 3-4 dias, até um máximo de 35 µg/Kg/dia) e/ou glucagom (1-10 µg/Kg/h em perfusão iv contínua, ou 0,02 mg/kg/dose iv em SOS). Nos casos refractários à terapêutica farmacológica, a intervenção cirúrgica é inevitável, sendo curativa na remoção das situações focais. Porém, no hiperinsulinismo difuso a abordagem é mais complexa, podendo requerer uma pancreatectomia de cerca de 95% do órgão com subsequentes défices enzimáticos e desenvolvimento de diabetes mellitus insulinodependente.
Na criança mais velha com hipoglicémia hiperinsulinémica, deve ser considerada a possibilidade da presença de insulinoma. Este pode ser uma entidade isolada, ou parte integrante da síndroma neoplásica endócrina múltipla tipo I (MEN I).
A síndroma de dumping ocorre após gastrectomia que, ao condicionar uma rápida entrada de hidratos de carbono no intestino delgado, provoca uma secreção excessiva de insulina, quer directamente, quer de forma indirecta através da activação de incretinas. Nos doentes com resistência à insulina, antes do desenvolvimento de hiperglicémia franca, podem ser segregadas grandes quantidades desta hormona após refeições ricas em hidratos de carbono, condicionando também episódios de hipoglicémia pós-prandial.
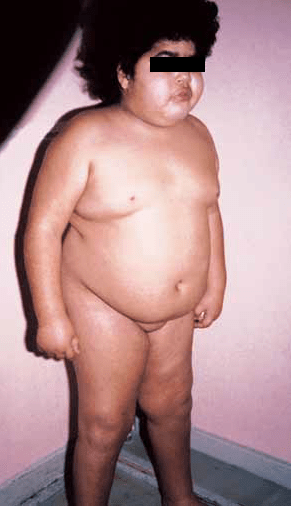
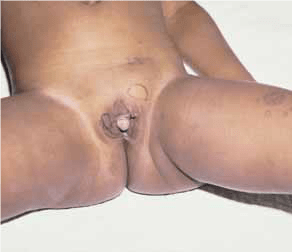
Até 50% dos doentes com síndroma de Beckwith-Wiedemann, assim como alguns defeitos da glicosilação (I-a, I-b e I-c), apresentam excesso de produção de insulina. A fisiopatologia do hiperinsulinismo ainda não está bem esclarecida, sendo o grau de hipoglicémia muito variável. Esta pode responder ao tratamento conservador, mas nalguns casos pode ser necessária pancreatectomia.
A hipoglicémica hiperinsulinémica também pode ser provocada pela administração de insulina. Esta situação distingue-se do aumento da produção endógena através da determinação do péptido-C plasmático, o qual se encontra anormalmente baixo perante insulinémia elevada.
Insuficiência adrenal
Como explicado previamente, o cortisol tem um papel importante na normalização glicémica, em situações de jejum e de estresse, através da estimulação da gliconeogénese e da libertação de adrenalina pela medula suprarrenal. Por outro lado, a sua deficiência amplifica a sensibilidade à insulina, com risco acrescido de hipoglicémia. A insuficiência adrenal pode ocorrer em qualquer idade, podendo ser originada por uma patologia que condicione disfunção primária da suprarrenal, por supressão da glândula secundária a corticóides, ou por insuficiência pituitária.
A doença de Addison é um processo auto-imune no qual o córtex suprarrenal é destruído por autoanticorpos, conduzindo à deficiência de gluco e mineralocorticóides, pelo que os doentes afectados podem apresentar hipoglicémia no contexto de doenças agudas intercorrentes. Tal afecção caracteriza-se por hiperpigmentação cutânea secundária à hiperestimulação dos melanócitos pelos elevados níveis de hormona adrenocorticotrópica (ACTH), que é produzida pela hipófise numa tentativa de estimular a produção de cortisol. Frequentemente, existe também hiponatrémia associada a hipercaliémia, apesar de a primeira poder surgir de forma isolada numa fase inicial da doença. Pode ainda haver associação a outras doenças auto-imunes como a diabetes mellitus tipo 1, o hipotiroidismo auto-imune, a doença celíaca ou a síndroma de poliendocrinopatia – candidíase – distrofia ectodérmica auto-imune (APECED). Esta última, herdada de forma autossómica recessiva, deve ser suspeitada na presença de dois dos três componentes cardinais: insuficiência adrenal auto-imune, hipoparatiroidismo auto-imune e candidíase. O diagnóstico é confirmado pela mutação do gene AIRE.
Outra causa de insuficiência adrenal é a síndroma de Allgrove ou síndroma dos três “a”: alacrimia, acalásia e insuficiência adrenal. Esta entidade é secundária a resistência à ACTH, apresentando disfunção autonómica e clínica neurológica progressivas. Com origem na mutação do gene ALADIN localizado no cromossoma 12q, é transmitida de forma autossómica dominante.
Em rapazes com insuficiência adrenal torna-se obrigatório excluir adrenoleucodistrofia, uma condição recessiva ligada ao cromossoma X, associada a níveis elevados de ácidos gordos de cadeia muito longa na urina. A insuficiência adrenal pode ser a primeira manifestação desta patologia, seguida de leucodistrofia do SNC, com deterioração neurológica progressiva.
Existe ainda uma forma muito mais rara, autossómica recessiva, que surge em idade mais precoce e com apresentação mais grave. A adrenomieloneuropatia é outra variação desta condição, com insuficiência adrenal na segunda infância e adolescência, seguida de alterações neurológicas 10 a 15 anos mais tarde. O óleo de Lorenzo não se mostrou eficaz na alteração do curso da doença, sendo o transplante de medula óssea o único tratamento capaz de prevenir a progressão da deterioração neurológica.
Outras causas raras de insuficiência adrenal congénita, com apresentação após o período neonatal, incluem a deficiência familiar de glucocorticóides (por resistência à ACTH) e a síndroma de Wolman (por deficiência da lípase ácida lisossómica). A hipoplasia congénita da suprarrenal habitualmente tem apresentação no recém-nascido, apesar de em alguns casos se tornar aparente apenas alguns meses mais tarde.
A disrupção do eixo hipotálamo-hipofisário-adrenal é também causa de insuficiência adrenal. Este défice de ACTH pode ser secundário a hipopituitarismo congénito, ou adquirido após tumores (como o craniofaringeoma), irradiação, traumatismos, ou infecções do SNC. O uso continuado de corticóides (incluindo os inalados) pode estar também na origem da supressão da ACTH hipofisária.
Défice de hormona do crescimento
À semelhança do cortisol, também a acção da hormona do crescimento (HC ou GH) tem maior importância durante o período de jejum prolongado, diminuindo a sensibilidade à insulina e a utilização da glicose, e estimulando a lipólise.
Recém-nascidos e pequenos lactentes com défice de HC podem apresentar hipoglicémia recorrente, com predomínio no início da manhã. Porém, o crescimento não é afectado antes do 1 ano de vida. Características importantes a ter em consideração na história clínica incluem a presença de: defeitos da linha média, micropénis e icterícia neonatal. Paralelamente ao que acontece com o défice de ACTH, o défice de HC pode ser isolado ou em contexto de hipopituitarismo, bem como congénito ou decorrente de agressão do SNC. Os doentes apresentam níveis séricos de IGF-1 inferiores ao normal para a idade e, por vezes, alterações evidenciadas através de ressonância magnética da sela turca. O diagnóstico deve ser confirmado através de provas de estimulação da HC.
Doenças hereditárias do metabolismo
Glicogenoses
As glicogenoses (GSD) são um conjunto de afecções caracterizadas essencialmente por defeitos enzimáticos que comprometem a síntese ou a degradação do glicogénio, com subsequente incapacidade de fornecimento de glicose em caso de necessidade.
Algumas atingem o fígado, o músculo e o coração, enquanto outras são específicas de órgão. A hipoglicémia ocorre na maioria das glicogenoses hepáticas, frequentemente acompanhada de hepatomegália. O diagnóstico é confirmado por estudo genético ou por estudo enzimático através de biópsias. Na maioria dos casos, o tratamento consiste na prevenção da hipoglicémia através de alimentação entérica contínua/cíclica e de amido de milho cru. (Quadro 3)
QUADRO 3 – Glicogenoses associadas a hipoglicémia
| (Adaptado de Langdon DR et al, Pediatric Endocrinology 2014) | |||||
| GSD TIPOS | Enzimas | Gene | Herança | Frequência da hipoglicémia | Gravidade da hipoglicémia em jejum |
| 0 | Glicogénio sintetase | GYS2 (12p12.2) | AR | Pouco frequente | Ligeira |
| Ia | Glicose 6-fosfatase | G6PC (17q21) | Frequente | Grave | |
| Ib | Glicose 6-fosfatase translocase | SLC37A4 (11q23) | Rara | Grave | |
| IIIa | Amilo-1,6-glucosidase hepática e muscular | AGL (1p21) | Frequente | Ligeira a moderada | |
| IIIb | Amilo-1,6-glucosidase hepática | Rara | Ligeira a moderada | ||
| IV | Enzima ramificadora | GBE (3p12.2) | Pouco frequente | Na insuficiência hepática avançada | |
| VI | Glicogénio fosforilase | PYGL (14q22.2) | Pouco frequente | Ligeira | |
| IXa | Subunidade α da cinase da fosforilase | PHKA2 (Xp22.13) | Ligada ao X | Frequente | Ligeira a moderada |
| IXb | Subunidade ß da cinase da fosforilase | PHKB (16q12.1) | AR | Rara | Ligeira a moderada |
| IXc | Subunidade γ da cinase da fosforilase | PHKG2 (16p11.2) | Rara | Ligeira | |
| XI | Transportador 2 da glicose | GLUT2 (3q26.2) | Rara | Ligeira | |
A GSD mais grave e com maior associação a hipoglicémia é a do tipo I ou doença de von Gierke. Sendo causada pela deficiência de glicose-6-fosfatase, impede a transformação da glicose-6-fosfato em glicose, a qual não pode ser libertada para a corrente sanguínea. Assim, o glicogénio acumula-se de forma contínua provocando hepatomegália maciça. Apesar de ser classificada como glicogenose, esta entidade é, primariamente, um defeito da gliconeogénese. Por este motivo, para além de hipoglicémia, estes doentes apresentam acidose láctica, hipertrigliceridémia e hiperuricémia.
A deficiência da amilo-1,6-glicosidase é responsável pela GSD tipo III ou doença de Cori-Forbes, com atingimento hepático e muscular secundariamente à incapacidade de desramificação do glicogénio. A sintomatologia traduz-se por hepatomegália franca (com aumento das transaminases) e baixa estatura, por vezes associadas a miopatia e cardiomiopatia graves. Contrariamente à GSD I, a hipoglicémia é menos frequente e menos grave, melhorando substancialmente após a puberdade. Por outro lado, uma vez que a gliconeogénese não é afectada, a acidose láctica, a hiperuricémia e a hipertrigliceridémia estão ausentes.
A GSD tipo IV ou doença de Andersen deve-se a uma deficiência da enzima ramificadora do glicogénio. Neste caso, a glicogenólise e a gliconeogénese não são afectadas, pelo que a hipoglicémia é rara na infância. Contudo, uma vez que ocorre lesão hepática progressiva, ela passa a ser mais frequente nos estádios avançados da doença.
A fosforilase é a enzima-chave da glicogenólise, necessitando de ser activada pela sua cinase, que é constituída por 4 subunidades. A deficiência da primeira enzima causa GSD tipo VI ou doença de Hers, enquanto a da segunda é responsável pela GSD tipo IX ou doença de Hug-Huijing. Ambas as condições têm manifestações semelhantes, com hepatomegália e aumento das transaminases secundárias à deposição de glicogénio. Existe ainda algum grau de restrição de crescimento, hipotonia e fraqueza muscular. A hipoglicémia é ligeira a moderada e associada a hipertrigliceridémia. A acidose láctica e a hiperuricémia estão ausentes. Em grande parte dos doentes, a clínica melhora após a puberdade, mas nalguns casos pode persistir miopatia, cardiomiopatia e acidose tubular renal.
A GSD tipo 0 é provocada pela deficiência de glicogénio-sintetase, pelo que não origina hepatomegália. Nestes casos, enquanto o jejum provoca hipoglicémia cetótica, após as refeições ocorre hiperglicémia com lactacidémia.
A síndroma de Fanconi-Bieckel ou GSD tipo XI é causada por mutações do gene GLUT2, que codifica o principal transportador de glicose da membrana celular do hepatócito. O movimento da glicose é essencial à sua homeostasia, pelo que defeitos do GLUT2 originam acumulação hepática de glicogénio com hepatomegália, bem como hiperglicémia pós-prandial e hipoglicémia de jejum. Esta última, acompanhada de cetose, resulta não só da diminuição do transporte do fígado para o sangue, mas também do excesso de perda urinária por diminuição da reabsorção no túbulo renal proximal. A fosfatúria que acompanha esta glicosúria pode ser suficientemente grave para provocar raquitismo hipofosfatémico, com subsequente hipocrescimento.
Defeitos da gliconeogénese
Com o prolongamento do jejum por mais de 12 a 16 horas, e já sem reservas de glicogénio hepático, a glicémia é reposta recorrendo à gliconeogénese. A alanina, o lactato e o glicerol são os principais substratos utilizados. A hipoglicémia recorrente e a acidose láctica são elementos comuns à deficiência das quatro enzimas da gliconeogénese, podendo ou não ser acompanhadas de cetose. Apenas o fígado e o rim possuem todas estas enzimas: o miocárdio e o tecido adiposo não apresentam frutose 1,6-bifosfatase, enquanto a glicose 6-fosfatase está ausente no músculo esquelético. A hipoglicémia grave e a hepatomegália são mais frequentes nos defeitos enzimáticos mais próximos da regeneração da glicose (frutose 1,6-bifosfatase e glicose 6-fosfatase), enquanto os que se encontram adjacentes ao ciclo de Krebs (piruvato carboxilase e fosfoenolpiruvato carboxicinase) estão mais associados a acidose láctica e a um processo neurodegenerativo progressivo. A apresentação, em regra, decorre no período neonatal.
Intolerância hereditária à frutose
Crianças com intolerância hereditária à frutose são assintomáticas até que esta seja introduzida na dieta. A deficiência de frutose-1,6 bifosfatase aldolase é herdada de forma autossómica recessiva, desencadeando hipoglicémia e dor abdominal sempre que é ingerida frutose. A sua evicção reverte automaticamente a situação, enquanto a sua persistência pode originar lesão hepática e renal, associadas a má evolução estaturoponderal.
Defeitos da oxidação dos ácidos gordos
A deficiência primária de carnitina é herdada de forma autossómica recessiva, provocando primariamente, como todos os defeitos da oxidação dos ácidos gordos e da cetogénese, hipoglicémia hipocetótica. Clinicamente, os doentes apresentam miopatia esquelética e/ou cardiomiopatia. A diminuição dos níveis plasmáticos de carnitina, particularmente durante episódios de hipoglicémia, deve levantar suspeita desta condição. Esta pode ser confirmada pela medição do transporte de carnitina nos fibroblastos (habitualmente inferior a 5% do normal).
O transporte da carnitina para a mitocôndria é realizado pelas carnitina palmitoiltransferase 1 (CPT1) e 2 (CPT2). A deficiência da primeira origina, não só hipoglicémia hipocetótica, mas também hepatomegália com aumento das transaminases, acidose tubular renal e miopatia, com elevação da creatinina-cinase. Existe ainda uma forma de apresentação mais grave, nos primeiros dias de vida, associada a convulsões e cardiomiopatia. O diagnóstico é confirmado pelo aumento dos ácidos dicarboxílicos de cadeia entre C6 e C10. A deficiência da CPT2 origina dois fenótipos:
– a forma benigna, na idade adulta, com início de rabdomiólise após exercício prolongado; – a forma grave, nos primeiros meses de vida, com envolvimento cardíaco e morte súbita antes do ano de idade. Tanto no défice primário como nas alterações do transporte, o tratamento consiste na evicção do jejum prolongado, tendo por base uma dieta pobre em gordura e enriquecida com triglicéridos de cadeia média e carnitina.
A deficiência da acil-CoA desidrogenase de cadeia média, herdada de forma autossómica recessiva, é o defeito mais frequente da oxidação mitocondrial dos ácidos gordos. A clínica surge habitualmente entre os 3 meses e os 3 anos, com hipoglicémia hipocetótica durante episódios de doença. Em caso de hipoglicémia grave, podem ocorrer convulsões com morbilidade a longo-prazo ou mesmo morte súbita. Com o avançar da idade e o aumento da tolerância ao jejum, os episódios de hipoglicémia tendem a melhorar. O diagnóstico é feito pela detecção urinária de octanoilcarnitina, fenilpropionilglicina e haxanoilglicina.
A acumulação de acilcarnitinas secundária à deficiência da acil-CoA desidrogenase de cadeia muito longa pode desencadear acidose láctica neonatal grave, cardiomiopatia e hepatopatia, de forma similar ao que acontece nos defeitos da cadeia respiratória mitocondrial. Variantes menos graves têm apresentação na adolescência, com atingimento dos músculos cardíaco e esquelético (com rabdomiólise, dor e fraqueza crónica).
Defeitos do metabolismo dos aminoácidos
A maioria das acidémias orgânicas que cursam com hipoglicémia (nomeadamente, a acidémia propiónica, e a doença urinária do xarope de ácer ou a acidúria 3-hidroxi-3-metilglutárica) tem apresentação no período neonatal. Contudo, nalgumas crianças com fenótipo menos grave, a clínica surge mais tarde, já com vários meses de vida. Ao contrário dos defeitos da oxidação dos ácidos gordos, estas alterações cursam com hipoglicémia hipercetótica, a qual se associa a outras manifestações como odores característicos, atraso de neurodesenvolvimento, hipotonia e acidose metabólica.
Exames complementares
Dada a enorme variedade de situações que podem desencadear hipoglicémia, tal circunstância gera habitualmente um verdadeiro desafio diagnóstico. Na verdade, em muitas crianças, mesmo após investigação clínica e laboratorial exaustiva, a etiologia não é identificada.
Uma necessidade de glicose superior a 8 mg/kg/min para manter a normoglicémia é, como foi referido antes, muito sugestiva de hiperinsulinismo. Outra informação valiosa a ter em consideração na anamnese é o tempo de jejum decorrido. Um episódio de hipoglicémia poucas horas após uma refeição aponta para excesso de insulina, apesar de tal facto também poder ocorrer em defeitos mitocondriais, ou após ingestão de frutose no doente com intolerância a esta pentose. Na glicogenose tipo I os episódios ocorrem após um breve período de jejum (1,5-4h), quando a glicose proveniente da dieta já foi consumida. Noutras doenças hereditárias do metabolismo, a hipoglicémia habitualmente tem lugar mais tardiamente: 4 a 12h noutras glicogenoses, 8 a 16h nos distúrbios da gliconeogénese, e após 10h nos defeitos da oxidação dos ácidos gordos.
Nalguns casos o exame objetivo leva à suspeita do diagnóstico: a hiperpigmentação pode ser sinal de insuficiência adrenal, enquanto a hepatomegália pode indicar glicogenose. Nos defeitos da oxidação dos ácidos gordos ou da gliconeogénese, o fígado está aumentado no episódio agudo, retornando ao normal posteriormente. A miopatia e a cardiomiopatia estão também presentes nalgumas DHM.
Todavia, na grande maioria dos doentes, o diagnóstico depende da investigação laboratorial (Quadro 4). As amostras de sangue, se possível, devem ser colhidas durante o episódio e antes da correcção da hipoglicémia (amostra crítica), mas sem atrasarem o tratamento. No caso de o volume de sangue ser limitado, deverão ser privilegiadas as determinações da glicose e da insulina. Por outro lado, os corpos cetónicos e os ácidos orgânicos deverão ser medidos na primeira urina após a correcção.
QUADRO 4 – Investigação laboratorial dos episódios de hipoglicémia
| Se possível, deverá ser congelada parte da amostra de sangue e de urina para eventual investigação futura. |
| (Adaptado de Ghosh A et al, Arch Dis Child 2016) |
|
Os glucómetros portáteis, habitualmente utilizados no controlo da diabetes mellitus, servem como método de rastreio para a determinação da glicémia e da cetonémia. Contudo, uma vez que os dispositivos existentes no mercado não são suficientemente precisos no diagnóstico de valores de glicémia inferiores a 60 mg/dL, é sempre necessária confirmação laboratorial. A concentração da glicose no sangue total é 10-15% inferior à do plasma, pelo que, por uma questão de consistência com os valores descritos na literatura, esta deverá ser medida no plasma. Outro factor a ter em consideração é a necessidade de celeridade no processamento da amostra, uma vez que a glicólise levada a cabo pelos eritrócitos poderá induzir um valor falsamente baixo.
Como foi explicado anteriormente, a presença de insulina durante um episódio de hipoglicémia é consistente com hiperinsulinismo. Todavia, esta pode não ser detectada numa amostra única, uma vez que sofre rápida depuração (clearance) hepática. Factores adicionais que corroboram este diagnóstico incluem: a diminuição plasmática dos ácidos gordos livres e dos corpos cetónicos (Figura 1); o incremento glicémico (> 25 mg/dL) em resposta à administração de glucagom ou de octreótido. Uma insulinémia elevada sem que haja o aumento correspondente do péptido-C plasmático deve levantar a suspeita de administração exógena. Por outro lado, a redução dos corpos cetónicos acompanhada do aumento dos ácidos gordos livres sugere a presença de um distúrbio da cetogénese ou da oxidação dos ácidos gordos. Esta última deve ser confirmada pelo doseamento da acilcarnitina

FIGURA 1. Diagnóstico diferencial de hipoglicémia
(Adaptado de Langdon DR et al, Pediatric Endocrinology 2014)
Abreviaturas: ß-OHB – ß-Hidroxibutirato; AGL – Ácidos Gordos Livres; GSD – Glicogenoses; HC – Hormona do Crescimento; SR – Suprarrenal; AG – Ácidos Gordos.
A inclusão do doseamento da hormona de crescimento e do cortisol na amostra plasmática inicial é controversa. Frequentemente, nas crianças sujeitas a provas de jejum, os níveis destas hormonas encontram-se abaixo do intervalo considerado normal, além de não se correlacionarem necessariamente com o grau de hipoglicémia. Se a suspeita for de insuficiência adrenal, o diagnóstico deverá ser estabelecido por uma prova de ACTH. Por outro lado, se recair sobre o défice da hormona de crescimento, além da avaliação antropométrica e das provas de estimulação da produção desta hormona, deve proceder-se a ressonância magnética da sela turca para avaliar a possível existência de alterações anatómicas hipotálamo-hipofisárias.
Apesar da importância da colheita da amostra crítica durante o episódio de hipoglicémia, actualmente várias condições metabólicas podem ser diagnosticadas de outra forma. Este avanço diagnóstico, aliado aos riscos inerentes às provas de jejum e à necessidade de uma monitorização muito rigorosa por uma equipa experiente, levou a que estas sejam executadas agora muito mais esporadicamente. A maioria dos defeitos da oxidação dos ácidos gordos pode ser identificada pela análise das acilcarnitinas no sangue, obtidas posteriormente com o doente já estabilizado. Se a clínica for sugestiva de glicogenose ou de alteração da gliconeogénese, o diagnóstico poderá ser confirmado pela sequenciação do painel dos genes mais relevantes. Contudo, os resultados genéticos devem ser interpretados de forma cuidadosa, uma vez que, se por um lado as mutações nem sempre são identificadas, por outro, podem ser encontradas variantes de significado incerto.
Tratamento
O tratamento de emergência da hipoglicémia sintomática consiste na pronta administração intravenosa de 2 a 5 mL/kg/dose de glicose a 10% (Quadro 5). O uso de soluções com concentração superior a 10% deve ser evitado, uma vez que estão descritos relatos de hiperglicémia grave por excesso de correcção. A glicémia deverá ser reavaliada 15 minutos depois, com repetição do bolus caso o valor não seja superior a 70 mg/dL. Deverá ser então iniciada uma perfusão de glicose a 10%, a um ritmo de 5 mL/kg/h (que providencia 8 mg/kg/minuto de glicose). Assim que a criança estiver consciente e com boa tolerância oral, a perfusão intravenosa poderá ser progressivamente descontinuada, mantendo a glicémia entre 70 mg/dL e 110 mg/dL.
QUADRO 5 – Tratamento de emergência da hipoglicémia
| (Adaptado de Ghosh A et al, Arch Dis Child 2016) |
Doente consciente e com tolerância oral
|
Doente inconsciente
|
| Doente inconsciente e sem acesso iv
Glucagom im: 0,5 mg se < 25 Kg; 1 mg se > 25 Kg |
Doentes com glicémia inicial < 50 mg/dL e/ou alteração do estado de consciência deverão ser internados para monitorização (inicialmente horária) e investigação diagnóstica.
A abordagem a médio e longo-prazo, bem como a prevenção de recorrências dependem essencialmente do correcto diagnóstico etiológico e tratamento específico. Antes da alta, é necessário assegurar que a criança consegue manter uma normoglicémia durante um período de jejum seguro, ou seja, durante o tempo habitual de sono adequado à sua idade (cerca de 4-6h no lactente; 12-16h na criança). Deve ainda ser fornecido glucómetro portátil e ser realizado ensino à família no que respeita à periodicidade e composição das refeições.
BIBLIOGRAFIA
Adamkin DH. Neonatal hypoglycemia. Semin Fetal Neonatal Med 2017; 22: 36 – 41
Butler G, Kirk J (eds). Paediatric Endocrinology and Diabetes. Oxford: Oxford University Press, 2011
Clarcke JTR (ed). A Clinical Guide to Inherited Metabolic Diseases. Cambridge: Cambridge University Press, 2004
Cryer PE. Williams Textbook of Endocrinology. Philadelphia: Elsevier, 2016
Davis SN, Lamos EM, Younk LM (eds). Endocrinology: Adult and Pediatric. Philadelphia: Elsevier, 2016
De Léon D, Thornton PS, Stanley SA, Sperling MA (eds). Pediatric Endocrinology. Philadelphia: Elsevier, 2014
Ghosh A, Banerjee I, Morris A. Recognition, assessment and management of hypoglycaemia in childhood. Arch Dis Child 2016; 101: 575–580
Güemes M, Rahman SA, Hussain K. What is a normal blood glucose? Arch Dis Child 2016; 101: 569–574
Güemes M, Hussain K. Hyperinsulinemic hypoglycemia. Pediatr Clin North Am 2015; 62: 1017-1036
Haddad NG, Eugster EA (eds). Endocrinology: Adult and Pediatric. Philadelphia: Elsevier, 2016
Hussain K (ed). Diagnostics of Endocrine Function in Children and Adolescents. Basel: Karger, 2011
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Long D, Akhtar Y. Hyperinsulinism. Pediatr Rev 2019; 40: 207-210; DOI: 10.1542/pir.2017-
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Radovick S, Misra M (eds). Pediatric Endocrinology. A Practical Clinical Guide. Berlin: Springer, 2018
Randell T. Diagnosis and management of hypoglycaemia beyond the neonatal period. Paed Child Health 2013; 23: 152-157
Shah R, McKinlay JD, Harding JE. Neonatal hypoglycemia: continuous glucose monitoring. Curr Opin Pediatr 2018; 30: 204-208
Sperling MA, Escobar O P-H (eds). Practical Algorithms in Pediatric Endocrinology Basel: Karger, 2016
Sunehag A, Haymond MW. Approach to hypoglicemia in infants and children. UpToDate. Disponível em: http://www.uptodate.com. (Acesso em Abril, 2017)
Thornton PS, Stanley CA, De Léon D, Harris D, Haymond MW, Hussain K, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of persistent hypoglycemia in neonates, infants, and children. J Pediatr 2015; 167: 238-245