Definição e importância do problema
As Esclerodermias Juvenis constituem um grupo de doenças raras do tecido conjuntivo que se traduzem globalmente no endurecimento e espessamento das camadas profundas da pele, com ou sem envolvimento visceral associado. Na idade pediátrica, as formas predominantes são as Esclerodermias Localizadas. A Esclerose Sistémica Juvenil (ESJ) é incomum. A distinção entre estas duas entidades é essencial, dadas as marcadas diferenças entre evolução clínica, abordagem terapêutica e prognóstico, mais grave e reservado nas formas sistémicas. As síndromas de sobreposição são mais comuns na criança que no adulto.
ESCLEROSE ou ESCLERODERMIA SISTÉMICA JUVENIL (ESJ)
A ESJ é uma doença crónica e multissistémica rara que causa fibrose cutânea, envolvimento de órgãos internos (esofágico, intestinal, cardíaco, pulmonar ou renal) e vasculopatia.
Aspectos epidemiológicos
O início juvenil é muito raro, sendo que as crianças e adolescentes com idade inferior a 16 anos constituem menos de 5% do total de casos de Esclerose Sistémica. O pico de incidência é entre os 10 e os 16 anos. A doença é quase 4 vezes mais frequente no sexo feminino, não havendo diferenças entre raças. A incidência anual estimada é 0,45 a 1,9 por 100.000 habitantes e a prevalência aproxima-se de 15 a 24 por 100.000.
Etiopatogénese
A causa da ESJ é desconhecida. A doença pode ser representada como um processo tripartido que origina um fenótipo predominantemente fibrótico. Neste estão envolvidos a disfunção do sistema imunitário (documentada pela presença de autoanticorpos específicos), a disfunção endotelial (associada à vasculopatia e fenómeno de Raynaud) e a disfunção dos fibroblastos (com aumento da síntese e deposição de proteínas da matriz extracelular e consequente fibrose). Estudos adicionais colocam a hipótese de alterações no tipo e metabolismo do colagénio, assim como na imunidade celular, estando aumentadas várias citocinas pró-inflamatórias (IL-1, IL-2, IL-4, IL-6 e IL-8).
Manifestações clínicas
As manifestações mais comuns na apresentação da ESJ são o fenómeno de Raynaud e a induração (esclerose) cutânea. O fenómeno de Raynaud é o sintoma mais frequente, estando presente em cerca de 70% dos casos no início da doença e pode preceder outras manifestações em muitos anos. A localização mais típica são os dedos das mãos, mas pode ser observado também nos pés, orelhas, lábios, língua e extremidade do nariz. Dez por cento dos casos complicam-se de úlceras digitais. As alterações cutâneas estão presentes em 40% dos casos na apresentação. Evoluem frequentemente em 3 estádios: edema (com duração de meses), induração que resulta no espessamento cutâneo com progressão proximal e nas contracturas (duração de 3 a 5 anos) e, por fim, atrofia (a pele torna-se esclerótica, fina e dura). Esta última fase é particularmente notória nos dedos e face, com a fácies característica que pode ser a primeira pista para o diagnóstico.
As Figuras 1 e 2 documentam sucintamente a tipologia das manifestações clínicas.
QUADRO 1 – ESJ: manifestações clínicas
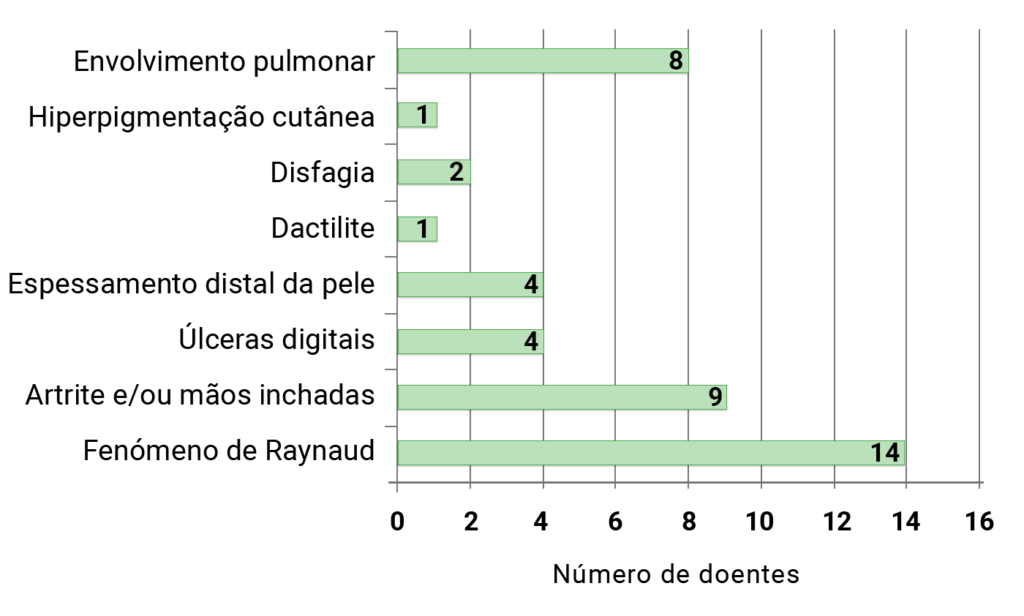
in Juvenile Systemic Sclerosis – Review of 15 patients. Sousa S, Fernandes S, Estanqueiro P, Zilhão C, Resende C, Ramos F, Salgado Mª Guedes M, Melo Gomes J, Santos MJ . Apresentado sob a forma de poster no congresso PRES, em Belgrado, Setembro 2014.
FIGURA 1. ESJ: manifestações clínicas
O envolvimento músculo-esquelético pode surgir no início ou com a evolução da doença sob a forma de artralgias, artrite, contracturas articulares (sobretudo nas articulações interfalângicas proximais e cotovelos), miosite (com mialgias, fraqueza muscular e elevação dos níveis de creatinocinase) ou calcinose subcutânea e periarticular. O envolvimento gastrintestinal pode ocorrer entre 30 a 70% das crianças e traduzir em disfunção esofágica (com queixas de refluxo gastro-esofágico e disfagia) e, mais raramente, alterações intestinais com alternância de padrão intestinal diarreia/obstipação, dor abdominal ou síndromas de malabsorção. O envolvimento pulmonar, com frequência assintomático, pode cursar com tosse seca e dispneia; ao contrário da ES do adulto, não cursa habitualmente com a devastadora fibrose intersticial. O envolvimento cardíaco é incomum mas é uma causa de morbilidade significativa nas crianças com ESJ (em caso de cardiomiopatia ou hipertensão pulmonar). O envolvimento renal, podendo ocorrer em 10% dos casos com proteinúria, hematúria ou insuficiência renal, habitualmente não é tão grave como nos adultos. As principais causas de morte na criança são as complicações cardiopulmonares.
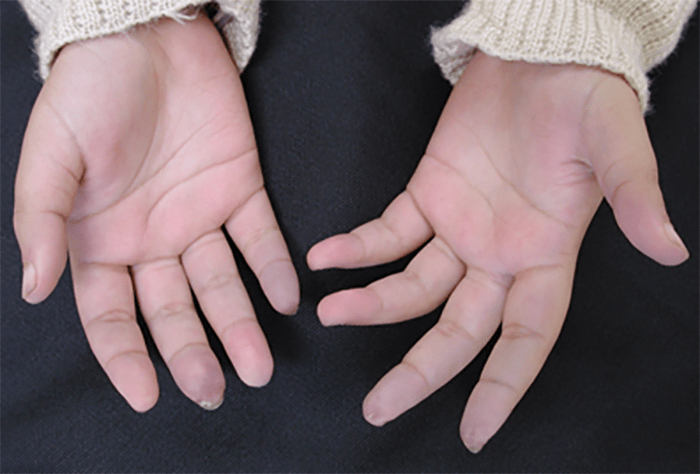
FIGURA 2. Fenómeno de Raynaud e lesões isquémicas das polpas digitais em jovem de 13 anos de idade com esclerodermia sistémica difusa iniciada cerca de 4 semanas antes. Note-se as lesões isquémicas já presentes ao nível das polpas digitais dos 3º e 5º dedos da mão direita e dos 2º e 3º dedos da mão esquerda, as quais regrediram completamente com a terapêutica instituída
Diagnóstico
O diagnóstico da ESJ é frequentemente atrasado pela raridade da doença. É estabelecido pela esclerose não confinada apenas a uma área (não localizada) acompanhada de envolvimento visceral e de auto-anticorpos. Os critérios seguintes foram elaborados pela Paediatric Rheumatology European Society (PRES), American College of Rheumatology (ACR) e European League Against Rheumatism (EULAR) em 2007. O diagnóstico de ESJ é feito se estiverem presentes o critério major + 2 dos 20 critérios minor. (Quadro 1)
A forma anteriormente considerada como “limitada” da doença geralmente apresenta Calcinose mais grave (que na forma difusa), fenómeno de Raynaud (que habitualmente complica com insuficiência vascular, ulcerações digitais e isquémia crítica levando a gangrena e por vezes necessidade de amputação de dedos), envolvimento Esofágico, eSclerodactilia e Telangiectasias, denominando-se por isso como Síndroma de CREST. Esta apresentação é muito rara na criança. Nos critérios de diagnóstico do PRES, ACR e EULAR, a forma limitada não surge como entidade distinta da forma difusa.
QUADRO 1 – Critérios de diagnóstico de ESJ
| Critério Major |
| · Esclerose cutânea proximal/induração da pele |
| Critérios Minor |
Pele
|
Vascular
|
Gastrintestinal
|
Renal
|
Cardíaco
|
Respiratório
|
Músculo-esquelético
|
Neurológico
|
Exames serológicos
|
Como resultado dos exames complementares verifica-se que a ESJ cursa com anemia de doença crónica (um quarto dos doentes), macrocitose (por má absorção), eosinofilia (15% dos doentes), leucocitose (relacionada com o grau de envolvimento visceral/muscular) e elevação de creatinocinase (se existir miosite). As crianças têm habitualmente títulos elevados de anticorpos antinucleares (ANA) (encontrados em 80-90% dos casos) mas a presença de anticorpos anticentrómero (em 7%) e anti-Scl 70 (em 34%) é muito menos frequente que nos adultos. A capilaroscopia do leito ungueal (CLU) é um exame não invasivo e fácil de realizar, fornecendo pistas importantes no diagnóstico e no seguimento dos doentes com ESJ. O padrão típico da capiraloscopia cursa com dilatações (precoce), dilatações e áreas avasculares (padrão activo) e tortuosidades e áreas avasculares (padrão tardio). A biópsia cutânea por vezes é importante para distinguir a ESJ de outras síndromas. Para avaliar o envolvimento esofágico – refluxo gastresofágico e diminuição da pressão no esfíncter esofágico inferior – está preconizada a realização de manometria e pHmetria de 24h. Para o rastreio e seguimento de envolvimento cardiopulmonar dever-se-á incluir radiografia de tórax (apesar de só permitir diagnosticar alterações consistentes com fibrose pulmonar numa fase avançada), TC torácica de alta resolução (demonstrando padrão em vidro despolido, “favo de mel”, opacidades lineares ou micronódulos subpleurais).
Provas de função respiratória com avaliação de difusão de monóxido de carbono (DLCO) que podem mostrar o padrão habitualmente restritivo com diminuição da DLCO e ecocardiograma transtorácico com Doppler e avaliação indirecta da pressão da artéria pulmonar para detectar hipertensão pulmonar, são outros exames a realizar em função do contexto clínico de cada caso.
Como suspeitar?
O diagnóstico precoce da ESJ é essencial dada a possibilidade de envolvimento de órgão que pode progredir rapidamente para complicações potencialmente fatais. Dada a raridade da doença e a subtileza que podem ter as manifestações iniciais, o diagnóstico é habitualmente atrasado em meses ou anos. O conhecimento do quadro clínico e elevado índice de suspeição podem ajudar no sentido de promover uma referenciação rápida para a Reumatologia Pediátrica.
A presença de fenómeno de Raynaud (mãos, pés ou outras localizações menos típicas), edema das mãos (“puffy hands”- mãos inchadas), úlceras digitais, diminuição da perfusão digital, atrofia da pele, contracturas de tecidos moles, calcinoses subcutâneas, fraqueza muscular, dispneia ou disfagia são sinais de alarme que deverão ser prontamente investigados.
Situações de crianças e adolescentes com fenómeno de Raynaud, padrão precoce ou activo típico na capilaroscopia do leito ungueal e presença de autoanticorpos específicos, incluídas numa forma “Pré-Esclerodermia”, deverão ter um seguimento rigoroso pelo risco de progressão para ESJ.
Tratamento
Dada a raridade da ESJ, pouca informação validada existe no que diz respeito ao tratamento. As medidas não farmacológicas incluem Medicina Física e Reabilitação e servem para ajudar a manter a função e a força musculares e ajudar a prevenir as contracturas. Outras medidas como protecção com luvas para o frio e hidratação da pele também são importantes.
Para o fenómeno de Raynaud, os bloqueadores do canal de cálcio são utilizados como fármacos de primeira linha (habitualmente a nifedipina). Nos casos graves de isquemia digital, com o Iloprost (prostanóide) demonstrou-se eficácia e segurança em crianças com ESJ, havendo alguma indicação para antagonista dos receptores da endotelina (Bosentan) em casos refractários de isquemia e úlceras digitais.
Para o envolvimento músculo-esquelético (artrite, miosite, tenossinovite) está indicada prednisolona em dose 0,3 a 0,5 mg/kg/dia (com o devido controlo da função renal e valores tensionais pelo risco de crise renal com a utilização de corticóides).
Para o envolvimento cutâneo e articular utiliza-se Metotrexato na dose de 15 mg/m2/semana, oral ou subcutâneo, em associação a corticoterapia. Para a ESJ com doença do interstício pulmonar, a Ciclofosfamida é normalmente considerada, em associação a corticoterapia.
Prognóstico
O prognóstico não é animador mas é melhor que nas formas do adulto. As contracturas articulares e o endurecimento cutâneo levam a morbilidade significativa. A taxa de mortalidade aos 5 anos é de 6 a 15%. As principais causas de morte nas crianças (em valores percentuais) são a cardiomiopatia (67%), a insuficiência renal terminal (13%), a insuficiência respiratória (10%), as infecções (7%), e a encefalopatia hipertensiva (3%).
ESCLERODERMIAS LOCALIZADAS JUVENIS (ELJ)
As ELJ englobam um variado número de quadros clínicos caracterizados pelo endurecimento cutâneo com aumento da deposição de colagénio. As lesões ao nível das estruturas anteriormente sistematizadas podem variar, de pequenas placas, até doença extensa com envolvimento de tecido celular subcutâneo, músculo e osso; como consequência, pode verificar-se evolução para deformidades funcionais e estéticas, mas sem envolvimento de órgãos internos. Na literatura dermatológica, os vários subtipos de EL também se denominam de “morfeia”. (Figuras 3 e 4)
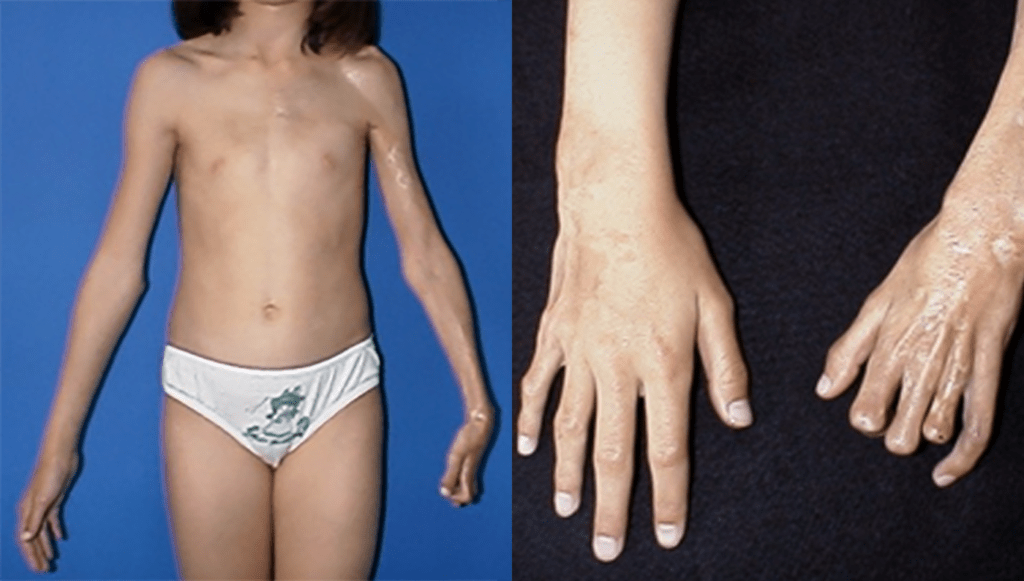
FIGURA 3. Jovem de 11 anos de idade com esclerodermia linear localizada, iniciada aos 7 anos de idade. São bem evidentes no membro superior esquerdo as lesões cutâneas escleróticas, com zonas de despigmentação e outras de hiperpigmentação, bem como a contractura digital devida às atrofias de partes moles da região palmar da mão esquerda, acompanhadas de melorreostose (atrofia do tecido ósseo adjacente)
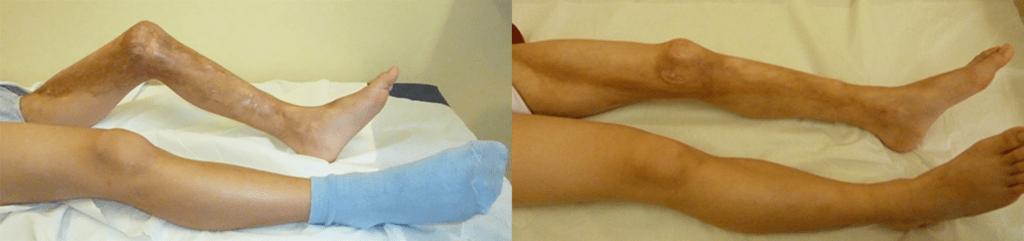
FIGURA 4. Esclerodermia linear do membro inferior esquerdo, em jovem de 9 anos de idade. Após 12 meses de terapêutica com corticóides e metotrexato subcutâneo, associada a fisioterapia adequada, conseguiu-se a melhoria marcada das lesões e correcção da contractura em flexão, mantendo uma adequada capacidade funcional (o jovem foi jogador de futsal durante todo este período)
Aspectos epidemiológicos
Apesar de incomum, a ELS é dez vezes mais comum que a ESJ. A incidência anual estima-se em 1 por milhão, com uma predominância do género feminino. O subtipo mais comum na criança é a Esclerodermia Linear, seguida da Esclerodermia em Placas. O pico de incidência verifica-se na idade escolar, com média nos 7,3 anos, havendo 6 descrições de casos no recém-nascido, todos com apresentação de Esclerodermia Linear (quatro deles “en coup de sabre”).
Etiopatogénese
A causa das ELJ é desconhecida. Há descrições de casos em associação com traumatismos locais, infecções víricas e bacterianas (Epstein-Barr e Borrelia burgdorferi) e vacinação (BCG e tríplice viral). Em termos histológicos, a esclerodermia é caracterizada por fibrose e espessamento de fibras de colagénio. Existe uma fase inflamatória inicial em que os feixes de colagénio da derme reticular se encontram espessados e há um infiltrado inflamatório intersticial e perivascular predominantemente linfocítico, ao nível da periferia das lesões. Na fase esclerótica tardia, ao nível do centro das lesões, há redução do infiltrado inflamatório e espessamento e agregação dos feixes de colagénio na derme, atrofia de glândulas exócrinas, sebáceas e folículos pilosos.
Manifestações clínicas
A doença é habitualmente autolimitada, com progressão variável consoante o subtipo clínico (mais rápida na Esclerodermia Linear dos membros, e mais gradual na Esclerodermia ou Morfeia em Placas). A ELJ divide-se em 5 subtipos: em placas ou circunscrita, linear (dos membros e da face/ “en coup de sabre”), generalizada, pansclerótica ou mista, sendo esta última uma combinação de dois ou mais subtipos dos anteriores. São apresentados de seguida os subtipos com base nos Critérios de Classificação de ELJ (Consensus Conference, Padua, 2004).
QUADRO 2 – Critérios de classificação da ELJ
| Esclerodermia em Placas |
| Superficial Áreas de induração redondas ou ovóides, únicas ou múltiplas, a nível da epiderme e derme, com halo violáceo à volta. A placa evoluiu infiltração nacarada e bordos mal definidos, de 2 a 15 cm, denominada de “iliac ring”. Localização preferencial nas cristas ilíaca, tórax, abdómen e membros inferiores. |
| Profunda ou Subcutânea Envolvimento subcutâneo e das camadas profundas da pele, com aspecto de “pele em casca de laranja” e, por vezes, ulceração. Localização preferencial no tórax superior e membros. |
| Esclerodermia Linear |
| Membros e Tronco Subtipo mais frequente em crianças e adolescentes, correspondendo a 60% dos casos e mais frequente em raparigas em idade escolar. Faixas lineares de esclerose que se podem estender à derme, tecido celular subcutâneo, músculos e ossos, com consequente deformidade. Habitualmente unilateral. |
| Face e Calote Craniana/ “En coup de sabre” Forma unilateral da face, de localização frontoparietal, com uma depressão central linear que se assemelha a um golpe de sabre e que pode levar a hemiatrofia da face. Pode acompanhar-se de complicações neurológicas (convulsões, cefaleia, hemiparésia e paralisia facial), oculares (uveíte e vasculite) ou problemas do maxilar/dentários. A síndroma de Parry-Romberg ou hemiatrofia facial progressiva cursa com alterações ósseas, musculares ou do tecido subcutâneo, com ou sem esclerose cutânea. (Figura 4) |
| Morfeia Generalizada |
| Subtipo raro. Induração da pele que se inicia na forma de Esclerodermia em Placas e que evolui para 4 ou mais lesões, com dimensões superiores a 3 cm, que se podem tornar confluentes e envolvem pelo menos 2 regiões anatómicas. Distingue-se da ESJ por não apresentar fenómeno de Raynaud nem alterações na capilaroscopia de leito ungueal. |
| Morfeia Pansclerótica |
| Subtipo extremamente raro mas muito grave. Envolvimento generalizado de toda a espessura da pele, tecido celular subcutâneo, músculo, osso a nível de tronco, extremidades, face e calote. Envolvimento de articular cursando com contracturas e retracções dos membros incapacitantes. |
| Esclerodermia Mista |
| Associação de subtipos. É frequente na criança, nomeadamente a associação entre a forma linear e em placas. |
O envolvimento extracutâneo na ELJ é sobretudo articular, neurológico, ocular e gastrointestinal. O envolvimento articular é o mais frequente (em 19%) e presente sobretudo nas crianças com Esclerodermia Linear (sob a forma de contracturas). As manifestações neurológicas estão presentes em 4% dos casos, nomeadamente na Esclerodermia Linear da face/ “en coup de sabre” e traduzem-se em epilepsia/crises convulsivas do tipo parciais complexas, cefaleias, alterações do comportamento e dificuldades na aprendizagem. Alterações evidenciadas através de ressonância magnética também foram documentadas (calcificações, anomalias na substância branca, malformações vasculares e vasculite do sistema nervosos central). O envolvimento ocular, presente em cerca de 3% dos casos, pode traduzir-se em alterações dos supracílios, uveíte anterior, episclerite, estrabismo, pseudopapiledema e distúrbios de refracção. As manifestações gastrintestinais ocorrem em cerca de 2% dos doentes e correspondem habitualmente a queixas de refluxo gastresofágico. (Figura 5)
Exames complementares
Procedendo a análises laboratoriais, pode observar-se eosinofilia nos doentes com Esclerodermia Linear e Generalizada, a qual se relaciona com a extensão da doença. Em cerca de metade dos casos graves verifica-se hipergamaglobulinémia IgM e IgG. A frequência com que são detectados ANA varia de estudo para estudo, (23% a 73%), sem correlação com o subtipo de doença; contudo, os referidos anticorpos /ANA são mais vezes encontrados na Esclerodermia Generalizada, seguindo-se a Esclerodermia Linear. Níveis elevados de anti-ss-DNA foram documentados na ELJ, sobretudo nos doentes com Esclerodermia Generalizada com envolvimento muscular; tal facto relaciona-se com a actividade e extensão da doença, assim como com a presença de contracturas articulares e deformidades. Os anticorpos anti-centrómero, anti-Scl 70, anti-Ro/la e anti-U1RNP surgem em 2-3% da ELJ, mas o seu significado prognóstico ainda é desconhecido. A presença de Factor Reumatóide correlaciona-se com a presença de artrite. A Termografia permite distinguir as lesões activas de inactivas (lesões novas ou activas têm uma diferença de +0,5ºC em relação à área adjacente ou membro contralateral); com uma sensibilidade de 92% e uma especificidade de 62%, poderá servir para o seguimento de Esclerodermia Linear. A Ressonância Magnética, permitindo visualizar as alterações estruturais e documentar a progressão da atrofia do tecido conjuntivo e ósseo, tem grande utilidade nos doentes com Esclerodermia Linear dos membros e “en coup de sabre” sobretudo na suspeita de envolvimento do sistema nervoso central. A Ecografia de alta frequência (20 MHz) permite determinar a profundidade e extensão da esclerose, assim como a perda de tecido adiposo subcutâneo e músculo. Outras técnicas em estudo são a Fluxometria por laser Doppler e a Imagem por laser Doppler.
Tratamento
A escolha do tratamento deve ter em conta a extensão, severidade e taxa de progressão da doença. O seu objectivo é prevenir o desenvolvimento de complicações funcionais e estéticas. A fisioterapia regular é essencial, sobretudo nos casos de Esclerodermia Linear para prevenir o desenvolvimento de contracturas e o prejuízo da capacidade funcional. Os corticóides tópicos estão indicados para a Esclerodermia em Placas (sobretudo se única) e fases precoces da doença, associados a hidratação diária da pele. Alguns estudos utilizaram a triancinolona injectada nas margens da lesão (com bons resultados apesar de aumentar o risco de necrose e lipodistrofia). Esta forma de ELJ responde ao tratamento tópico com corticóides, análogos de vitamina D3 e/ou fototerapia. A terapia com PUVA, indicada para os estádios iniciais da inflamação, assim como a terapia com UVA 1, são mais benéficas nos estádios de fibrose. O calcipotriol 0,005% tópico em associação a UVA1 leva à redução do espessamento e hiperpigmentação cutâneas. O tratamento com imiquimod, imunossupressor utilizado para tratamento de quelóides e carcinoma espino e basocelular, também se mostra promissor na ELJ. Os corticóides orais são utilizados quando existe exuberante inflamação local, rápida progressão da lesão, prejuízo funcional, risco de interferência no crescimento e presença de lesão muscular. O metotrexato subcutâneo (15-20 mg/m2/semana) tem sido utilizado com benefício na ELJ grave que afecta tecido celular subcutâneo, músculo, fáscia e/ou ossos (nomeadamente formas de Esclerodermia Linear e Generalizada); por vezes tem sido associado a pulsoterapia mensal com metilprednisolona (500 mg/m2), reduzindo o espessamento cutâneo. A associação de metotrexato e corticoterapia também se mostrou eficaz como primeira linha nos casos graves de ELJ. Em casos de doença refractária, a cirurgia poderá ser equacionada.
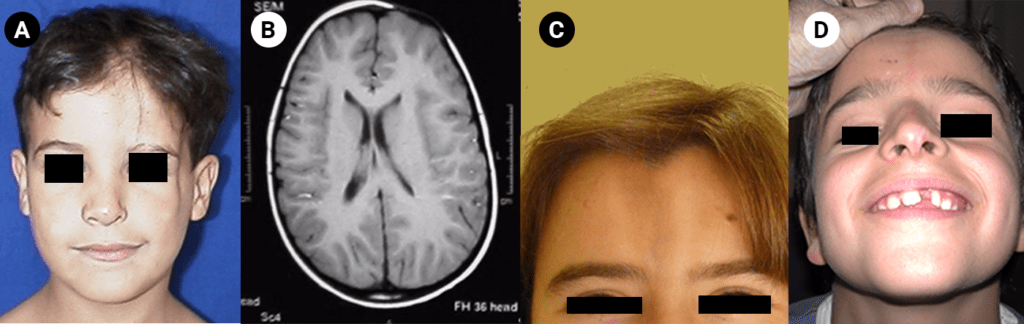
FIGURA 5. Síndroma de Parry-Romberg – (A) note-se a assimetria facial e em (B) note-se a zona de alteração da medular do frontal à esquerda; Eslerodermia Linear “en coup-de-sabre” em dois doentes diferentes: em (C) é visível uma lesão bilateral e em (D) note-se o a atrofia da aba nasal e o hipodesenvolvimento do 1º incisivo esquerdo, localizados no mesmo meridiano da lesão da região frontal
Prognóstico
A ELJ não evolui para ESJ, sendo a sobrevida sobreponível à da população geral. As formas de Esclerodermia Linear apresentam potencialmente maior risco de incapacidade física e deformidades estéticas. Na apresentação segmentar dos membros inferiores, em cerca de 20% das crianças desenvolve-se atrofia significativa dos tecidos adjacentes, podendo levar a uma diferença até cm no comprimento dos membros. Na apresentação de tipo “en coup de sabre” pode haver uma evolução para atrofia óssea da calote, deformidade do maxilar superior, posições anormais da dentição e envolvimento do sistema nervoso central.
BIBLIOGRAFIA
Charles C, Clements P, Furst DE. Systemic sclerosis: hypothesis – driven treatment strategies. Lancet 2006; 367: 1683-1690
Goldman L, Schafer AI (eds). Goldman – Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Jolles S, Stein MR, Longhurst HJ, et al. New frontiers in subcutaneous immunoglobulin treatment. Biologics in Therapy 2011; 1: 3-6
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Li SC. Scleroderma in children and adolescents: localized scleroderma and systemic sclerosis. Pediatr Clin North Am 2018; 65: 757-782
Macedo PA, Shinjo S, Goldenstein-Schainberg C. Esclerodermia Juvenil. Acta Reumatol Port 2008; 33: 289-297
Morel Z, Benadón E, Faugier E, Maldonado MR. Características clínicas de niños com esclerodermia en un hospital de referencia. Reumatol Clin 2009; 5: 158-162
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Reed AM, Manson II TG (eds). Pediatric Rheumatology- a Color Handbook. London: Manson Publishing, 2012
Silva M, Knoll Palma L, Knob C et al. Esclerose Sistémica Juvenil: uma doença incomum na infância. Acta Pediatr Port 2012; 43: 257-259
Tashkin DP, Elashoff R, Clements P, et al. Cyclophosphamide versus placebo in scleroderma lung disease. NEJM 2006; 354: 2655-2666
Vehe RK, Riskalla MM. Collagen vascular diseases: SLE, Dermatomyositis, Scleroderma, and MCTD. Pediatr Rev 2018; 39: 501-513
Wigley FM. Raynaud’s phenomenon. NEJM 2002; 347: 1001-1008
Zancanaro PC, Garcia LT, Isaac AR, Costa IM. Localized scleroderma in children: clinical, diagnostic and therapeutic aspects. An Bras Dermatol 2009; 84:161-72
Zulian FF, Atheya BH, Laxer et al in behalf of Juvenile Scleroderma Working Group of the Pediatric Rheumatology European Society (PRES). Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford) 2006; 45: 614-20.
Zulian F, Vallongo C, Woo P, et al. Localized scleroderma childhood is not just a skin disease. Arthritis Rheum 2005; 52: 2873-2881