Etiopatogénese e generalidades
A hiperplasia congénita da suprarrenal (HCSR) é uma expressão que engloba um grupo de doenças autossómicas recessivas, causadas por défices enzimáticos da via de síntese de esteróides ou, muito raramente, por défice da proteína P450 oxirredutase (POR) interveniente em vários passos da síntese metabólica envolvendo transferência de electrões para o citocrómio P450 microssomal. (Figura 1 do capítulo anterior)
A perda do retrocontrolo negativo do eixo hipotálamo-hipófise-suprarrenal e o défice de cortisol levam à secreção aumentada e estimulação permanente da ACTH, com consequente hiperplasia do córtex da SR e hiperprodução de metabolitos intermediários.
O défice mais frequente é o défice em 21-hidroxilase (destacada neste capítulo), responsável por 90 a 95% dos casos; todas as outras formas de hiperplasia são relativamente raras. Assim, é dada ênfase a esta forma, apresentando-se no Quadro 1 e de forma resumida, as principais características clínicas e laboratoriais de outros défices enzimáticos.
HCSR por DÉFICE de 21-HIDROXILASE
Etiopatogénese e epidemiologia
A incidência desta doença é variável consoante a população estudada: 1/280 no Alasca e 1/23.000 em França; considerando apenas as formas graves do recém-nascido e criança obtém-se valor de 1/14.000; incluindo as formas de apresentação tardia a taxa será ~1/1.000 nos indivíduos em geral e 1/27 nos judeus Ashkenazi.
Nos países da Europa e nos EUA, com programas de diagnóstico precoce, é possível identificar formas graves e algumas das formas mais ligeiras, sendo a respectiva incidência, também variável, oscilando entre 1/15 e 1/16.000.
Os 2 genes (CYP21P e CYP21) que codificam a 21-hidroxilase estão localizados no braço curto do cromossoma 6, muito perto dos locus dos HLA e C4; só um dos genes é activo, sendo o outro um pseudogene; a maioria dos doentes corresponde a duplos heterozigotos, tendo herdado duas mutações diferentes, sendo a gravidade do quadro clínico determinada pela mutação mais suave. A grande variedade de mutações associadas, determinando graus muito variáveis de função enzimática, origina formas clínicas de gravidade variável.
QUADRO 1 – Formas de hiperplasia congénita da suprarrenal**
| Δ4A: Δ4-androstenediona; DHEA: de-hidroepiandrosterona; DHEA-S: sulfato de de-hidroepiandrosterona; DOC: desoxicorticosterona; F: feminino; HTA: hipertensão arterial; M:masculino PRA: actividade da renina plasmática; T: testosterona; PA: pressão arterial; * StAR é a designação da proteína de regulação aguda da esteroidogénese (Steroidogenesis Acute Regulatory protein), essencial para o transporte de colesterol do citoplasma da célula do córtex suprarrenal para a mitocôndria onde ocorrem alguns dos passos da síntese hormonal; ** Excluindo o défice de 21-hidroxilase, abordado no texto. | ||||||||
| Défice | Síndroma (Frequência) | Ambiguidade genital | Virilização pós-natal Puberdade | Metabolismo do sal | Crise aguda PA | Esteróides ↑ ≠ | Esteróides ↓ | Localização do gene |
Proteína P450 | Fenótipo variável por vezes associada a malformações semelhantes ao S. Antley-Bixler | Sim F e M | Não | Não | Não | 17-OH-progesterona | DHEA, D4A e T | 7q |
| Proteína StAR* | Hiperplasia lipóide (Rara) | Marcada no sexo M | Não Ausência de puberdade no sexo F | Perda de sal de aparecimento tardio ↑ PRA | Frequente PA ↓ | Nenhum | Todos | 8p |
| 3β OH – Esteróide (Rara) desidrogenase | Clássica (Rara) | Marcada no sexo M Moderada no sexo F | Sim Alterações da puberdade | Perda de sal ↑ PRA | Presente PA ↓ | DHEA, DHEA-S, 17OH – pregnenolona | Aldosterona, T, cortisol | 1p |
| Não clássica (Frequente) | Não | Sim Alterações da puberdade | Normal | Ausente PA normal | DHEA, DHEA-S, 17OH – pregnenolona | …. | ? | |
| 17α – Hidroxilase | (Rara) | Sexo M | Não Ausência da puberdade no sexo F | ↑ PRA | Ausente HTA | DOC, corticosterona | Cortisol, T, DHEA, Δ4A, 17OHP, 17OH – pregnenolona | 10q |
| 11β – Hidroxilase | Clássica (1 / 100.000) | Sexo F | Sim | ↑ PRA | Rara HTA | 11 – desoxicortisol DOC, T, Δ4A | Cortisol± aldosterona | 8q |
| Não clássica (Frequente?) | Sexo F | Sim Alterações da puberdade | Normal | Ausente PA normal | 11 – desoxicortisol ± DOC, T, Δ4A | 8q | ||
| Corticosterona metiloxidase II | Perda de sal (Rara) | Não | Não | Perda de sal | Só perda de sal | 18OH – corticosterona | Aldosterona | 8q |
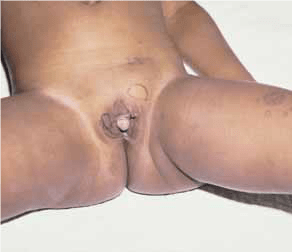
FIGURA 1. Ambiguidade genital no contexto de HCSR
A actividade das suprarrenais inicia-se na vida intrauterina. Após o nascimento, a produção de cortisol e de aldosterona passa a ser de primordial importância para a sobrevivência.
Habitualmente, considera-se existirem duas formas de apresentação clínica e gravidade diferentes – a forma clássica virilizante com ou sem perda de sal, e a forma não clássica sem perda de sal; no entanto, esta classificação é artificial e arbitrária, considerando-se que existe um espectro contínuo de formas clínicas desde muito graves até assintomáticas.
1. Forma clássica virilizante, com ou sem perda de sal
Manifestações clínicas
Durante a gestação, a produção deficiente de cortisol e aldosterona não produz sintomas, mas a hiperestimulação mantida pela ACTH com produção excessiva de androgénios leva a virilização que pode ter graus variáveis: no sexo feminino poderá chegar à ambiguidade genital completa (genitais externos com aspecto “de rapaz” mas com criptorquidia bilateral e hipospadia); no sexo masculino poderá manifestar-se como macrogenitossomia e escroto hiperpigmentado, não chamando geralmente a atenção.
A Figura 1 mostra uma imagem de ambiguidade genital em criança de quatro anos.
No sexo masculino, a virilização poderá somente ser detectada mais tarde, geralmente entre os 3 e os 7 anos, por aceleração do crescimento e maturação óssea desencadeada pelos níveis elevados de androgénios suprarrenais (puberdade precoce periférica ou pseudopuberdade precoce). Tal situação pode mesmo desencadear uma puberdade precoce central após o início da terâpeutica. Em 75% dos casos de virilização existe perda concomitante de sal.
Os sintomas de insuficiência suprarrenal instalam-se lentamente durante as primeiras semanas de vida com má progressão ponderal, recusa alimentar, mau estado geral, desnutrição, vómitos, obstipação; os sintomas podem ainda instalar-se de forma aguda, gravíssima, com letargia, choro fraco, prostração, vómitos, diarreia, desidratação, hipotensão, acompanhados de hiponatrémia, hipercaliémia, hipoglicémia e acidose metabólica. Este quadro, se não for diagnosticado e tratado rapida e correctamente, pode mesmo conduzir ao colapso cardiocirculatório e morte. Esta é a forma de apresentação mais frequente no sexo masculino.
O diagnóstico diferencial da crise aguda de perda de sal faz-se com sépsis, gastrenterite e, sobretudo, com a estenose hipertrófica do piloro cujas manifestações clínicas são semelhantes. No entanto, nesta última situação surge hiponatrémia, hipocaliémia e alcalose metabólica.
Nas formas com virilização tardia, o exame objectivo mostra: estatura elevada com aceleração da velocidade de crescimento (haverá que comparar com a curva de crescimento da criança, se existirem dados anteriores), pilosidade púbica e/ou axilar (que se quantifica pelos estádios de Tanner), sudação com odor, e acne; no sexo masculino: pénis bem desenvolvido, testículos pequenos; no sexo feminino: clitoromegália e ausência de botão mamário. A presença de botão mamário e o aumento do volume testicular farão suspeitar puberdade precoce central desencadeada pelo avanço da idade óssea associado à hiperplasia congénita da suprarrenal. A hiperpigmentação é habitualmente mais marcada nas palmas das mãos, pregas de flexão, cicatrizes, mucosa oral, mamilos, escroto e grandes lábios.
Exames complementares
Os exames complementares de diagnóstico que apoiam a suspeita clínica de crise aguda com perda de sal são:
- Ionograma plasmático: [diminuição de Na+], [elevação do K+];
- pH e gases: acidose metabólica;
- Glicémia: < 40 mg/dL;
- Ureia: elevada;
- Aumento dos níveis séricos de 17OH-progesterona, D4-androstenediona, testosterona, da actividade de renina plasmática (PRA) ou renina activa.
Nas formas graves os doseamentos basais são geralmente suficientes e os únicos que é possível obter face à emergência clínica do tratamento.
Para confirmação de forma virilizante não completamente esclarecida, pode ser necessário proceder à prova de estimulação com ACTH (Synacthen®) e determinação de 17OH-progesterona, D4-androstenediona, testosterona, DHEA, e 3-beta-desoxicortisol basais e 60 minutos após administração.
Os níveis séricos dos vários metabolitos variam de acordo com a idade e sexo da criança e, também, com os valores de referência de cada laboratório; por isso, devem utilizar-se para a interpretação dos resultados as tabelas locais de referência. A colheita de sangue para estes doseamentos deve ser efectuada entre as 8 e as 9 horas para evitar resultados falsamente negativos causados pela variação circadiana dos seus níveis plasmáticos.
Tratamento
1. De substituição
Glucocorticóide
O tratamento é, ainda hoje, uma das áreas que suscita mais discussão; tem por objectivo a administração de corticóide suficiente para frenar a produção de androgénios sem, no entanto, afectar o crescimento. Deve, assim, ser ajustada ao doente e alterada ao longo do tempo, de acordo com os dados clínicos (velocidade de crescimento, hiperpigmentação, pilosidade, genitais externos) e os níveis séricos dos metabolitos. Esta avaliação regular deverá ser trimestral nos 2 primeiros anos de vida podendo, depois, passar a semestral até à puberdade, altura em que os ajustamentos deverão voltar a ser trimestrais.
Nas crianças utiliza-se: hidrocortisona per os em doses de 10-15 mg/m2/dia dividida em 3 tomas, de preferência; nos recém-nascidos e lactentes administra-se 1,5 mg, 3 vezes por dia (papéis manipulados na farmácia).
Nas idades pós-fase de crescimento poder-se-á passar a dexametasona: 0,25-0,5 mg per os, à noite.
Mineralocorticóide
- 9 alfa-fludrocortisona:
- 50-300 µg/dia, per os, em 2 tomas nos lactentes;
- 50-200 µg/dia, per os, em 2 tomas nas crianças.
NaCl: 1-3 g/dia per os nos lactentes
As crianças maiores não necessitam de doses tão elevadas de mineralocorticóides; após o período de lactente, os alimentos que as crianças recebem contêm maiores concentrações de sal e as mesmas desenvolvem também apetência por sal, pelo que não é necessário administrar já NaCl suplementar.
2. Em situações de estresse agudo
Os doentes com hiperplasia congénita da suprarrenal deverão ter sempre consigo uma informação acerca da sua situação clínica, terapêutica actualizada e indicações em caso de estresse.
A família deverá ser informada que em situações de estresse agudo (como febre alta > 38,5ºC, vómitos, diarreia, administração de vacinas nos lactentes), deverá duplicar-se ou triplicar-se a dose habitual de hidrocortisona, de acordo com a gravidade do caso. Este tratamento deverá ser mantido pelo menos durante 24 horas; os pais devem também estar instruídos sobre a importância de administrar à criança líquidos/alimentos açucarados em quantidade suficiente para evitar a hipoglicémia que pode acompanhar estas situações, em particular nos lactentes e crianças mais jovens.
Se a criança vomitar imediatamente após a administração da terapêutica, dever-se-á repetir a dose algum tempo depois; se não tolerar a terapêutica por via oral, deverá ser rapidamente encaminhada a um serviço de urgência.
3. Da crise aguda de perda de sal
Deverá ser instituída terapêutica com fluidos por via endovenosa. Deverá ser instituída imediatamente terapêutica com hidrocortisona em bólus IM ou IV (o que for mais fácil e rápido): < 1 ano ” 25 mg; 1-5 anos ” 50 mg; > 5 anos ” 100 mg mantendo-se depois estas doses IV de 6/6 horas até reversão do quadro clínico, continuando terapêutica com fluidos IV.
Dever-se-á colher sangue para a determinação de glicémia, ionograma, pH e gases.
Se existir choque: NaCl a 0,9% a administrar ao ritmo de 20 ml/kg/h.
Se ausência de choque: NaCl a 0,9% em dextrose a 5% a administrar de acordo com os cálculos para a manutenção + perdas calculadas.
Não é necessário administrar mineralocorticóides, pois as doses altas de hidrocortisona asseguram estas necessidades.
Há que manter a hidratação endovenosa enquanto necessário. Introduzir líquidos per os logo que possível. Passar a terapêutica oral com hidrocortisona em dose tripla e fludrocortisona o mais brevemente possível; manter estas doses altas enquanto se mantiver o estresse agudo.
4. Da ambiguidade sexual
A atitude de proceder à intervenção cirúrgica para correção do defeito, respeitando os princípios éticos e a opinião esclarecida dos pais/família, deverá ser realizada por cirurgião pediátrico e equipa cirúrgica com experiência nestas situações. Habitualmente opta-se pela feminização.
2. Forma não clássica sem perda de sal
Manifestações clínicas
Esta forma de apresentação corresponde, em geral, a formas menos graves de défice enzimático. A hiperestimulação da glândula pela ACTH permite manter níveis plasmáticos normais de cortisol, à custa do aumento dos níveis plasmáticos dos precursores.
Manifesta-se tardiamente por pubarca precoce (aparecimento de pilosidade púbica e/ou axilar antes dos 8 anos no sexo feminino e 9 anos no sexo masculino), virilização, hirsutismo peripubertário, acne quística, amenorreia ou alterações menstruais, e infertilidade na mulher adulta; pode mesmo não haver quaisquer sintomas, sendo esta forma clínica diagnosticada por investigação da família no âmbito do diagnóstico de um caso índice.
Esta forma não se acompanha de perda de sal clinicamente significativa.
Exames complementares
- Radiografia da mão e punho esquerdos para avaliação da idade óssea:
- Idade óssea avançada em relação à idade cronológica
- Análises de sangue:
- Ionograma normal
- Níveis plasmáticos basais de 17OH-progesterona elevados. No sexo feminino e após a menarca, este doseamento deve ser realizado durante a fase folicular do ciclo
- Testosterona, D4-androstenediona, DHEA e DHEA-S: valores variáveis e sobreponíveis a outras situações
- PRA ou renina activa normais ou ligeiramente aumentadas
- Prova de estimulação com ACTH* (Synacthen®) se os valores basais dos parâmetros hormonais atrás referidos não forem conclusivos
| *Recorda-se a prova de ACTH sintética: medição do incremento do cortisol plasmático 30 minutos e 60 minutos após injecção IM de 0,25 mg de β1-24-corticotrofina. |
Tratamento
Têm indicação para tratamento os casos em que existe: avanço da idade óssea com prognóstico estatural inferior à altura alvo da família; hirsutismo importante; acne grave; alterações menstruais; massas testiculares.
A terapêutica é semelhante à descrita atrás: hidrocortisona per os em doses de 10-15 mg/m2/dia, dividida em 3 tomas. Não é necessário habitualmente proceder a substituição mineralocorticóide.
Nos doentes em tratamento e em situação de estresse agudo dever-se-á também duplicar ou triplicar as doses.
Os casos assintomáticos, sem indicação para terapêutica continuada com hidrocortisona, deverão também ser submetidos a terapêutica em caso de estresse (40-60 mg/m2/dia) se a resposta do cortisol na prova de Synacthen for inferior a 18 ug/dl.
BIBLIOGRAFIA
Brook C, Clayton P, Brown R (eds). Brook’s Clinical Pediatric Endocrinology. London: Blackwell Publishing, 2009
Claahsen-van der Grinten HL, Stikkelbroeck NMML, Otten BJ, Hermus ARKMM. Congenital adrenal hyerplasia- pharmacologic interventions from the prenatal phase to adulthood. Parmacol & Therapeutics 2011; 132: 1-14
Clayton P, Oberfield S, Ritzen E, et al. Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Pediatric Endocrinology. J Clin Endocrinol Metab 2002; 87: 4048-4053
Friães A, Rego AT, Aragues JM, et al. CYP21A2 mutations in Portuguese patients with congenital adrenal hyperplasia: Identification of two novel mutations and characterization of four different partial gene conversions. Mol Gen Metabol 2006; 88: 58-65
Hindmarsh PC. Management of the child with congenital adrenal hyperplasia. best Pract & Res Clin Endocrinol Metabol 2009; 23: 193-2008
Hughes IA. Congenital adrenal hyperplasia: a lifelong disorder. Horm Res 68 (suppl 5): 84-89, 2007
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet 2005; 365: 2125-2136
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
New M, Abraham M, Gonzalez B et al. Genotype-phenotype correlation in 1507 families with congenital adrenal hyperplasia owing to 21-hydroxylaser deficiency. PNAS 2013; 110: 2611
Nimkarn S, Lin-Su K, New MI. Steroid 21 hydroxylase deficiency congenital adrenal hyperplasia. Endocrinol Metab Clin Am 2009; 38: 699-718
Reisch N, Arlt W, Krone N. Health problems in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res Paediatr 2011; 76: 73-77
Speiser PW, Azziz R, Baskin LS et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2010; 95: 4133-4160
van der Kamp Hj, Wit JM. Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol 2004; 151: U71-U75
White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Res 2000; 21: 245-291