Definição e aspectos epidemiológicos
O linfoma de Hodgkin, tradicionalmente designado como doença de Hodgkin, designação abandonada pela OMS, tem o nome do médico que primeiro o descreveu, no início do século XIX. Trata-se dum processo maligno do sistema linforreticular de localização ganglionar, evidenciando escassez de células neoplásicas no gânglio afectado; neste, predomina uma população maioritariamente (90-99%) de células reactivas não neoplásicas, linfócitos, macrófagos e eosinófilos.
Constituindo cerca de 5% dos casos de cancro em idade pediátrica, surge nos países industrializados com um primeiro pico de incidência por volta dos vinte anos, e outro, a partir dos cinquenta anos. Raro antes dos 7 anos de idade, ao contrário dos outros linfomas, surge principalmente na pré-adolescência ou adolescência.
As relações entre esta doença e o vírus de Epstein Barr são conhecidas, já que, como foi referido anteriormente, o genoma do vírus se encontra com grande frequência na célula neoplásica.
O linfoma de Hodgkin é mais frequente em crianças com imunodeficiência, tendo sido descrito o seu aparecimento em “epidemias” familiares, porventura relacionadas com infecções víricas.
Manifestações clínicas
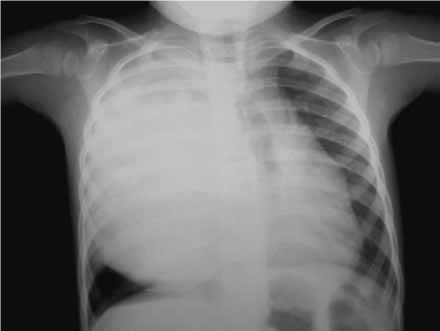
FIGURA 1. Linfoma de Hodgkin: opacidade esferóide “gigante” com ponto de partida mediastínico ocupando o campo pulmonar direito (NIHDE).
Clinicamente, caracteriza-se por ter um início insidioso, com aparecimento de adenomegálias principalmente cervicais, supraclaviculares ou mediastínicas. Mais raramente, a localização é infradiafragmática. Os gânglios são elásticos, não dolorosos, sem sinais inflamatórios, de crescimento muito lento. Por vezes, apresentam regressão espontânea durante algum tempo, no que o linfoma de Hodgkin se distingue da generalidade dos outros linfomas. Em fases mais avançadas, as adenomegálias podem confluir, formando conglomerados mais ou menos volumosos.
As adenomegálias do mediastino são assintomáticas de início, podendo ser um achado ocasional em radiografia. Mais tarde, os gânglios comprimem as estruturas vizinhas, podendo originar dispneia, disfagia e rouquidão (Figura 1). A progressão da doença faz-se, em regra, por via linfática, atingindo sucessivamente os territórios ganglionares vizinhos.
A presença de várias manifestações sistémicas (temperatura superior a 38ºC durante pelo menos três dias, emagrecimento superior a 10% do peso nos últimos seis meses, prurido e sudação nocturna), está associada a prognóstico mais reservado, o que obriga a terapêutica mais intensiva.
Diagnóstico
O diagnóstico é feito por biópsia de um gânglio que revela as células de Reed-Sternberg e suas variantes, num fundo de células inflamatórias (linfócitos, plasmócitos, eosinófilos, histiócitos), com fibrose. As células de Reed-Sternberg, de origem desconhecida durante muito tempo, foram identificadas imunologicamente como células linfóides de linhagem B, englobando-se esta doença actualmente no grupo dos linfomas.
As referidas células têm grande diâmetro (15-45 µm) e são multinucleadas ou com núcleo multilobulado. Reiterando o que foi referido antes, no linfoma de Hodgkin, ao contrário de outras neoplasias, as células neoplásicas correspondem a não mais do que 1% das células que se encontram nos gânglios atingidos, sendo as restantes células inflamatórias; tal particularidade poderá dificultar o diagnóstico.
De acordo com as células predominantes no gânglio e o grau de fibrose, classifica-se o linfoma de Hodgkin em: – com predomínio linfocitário; – com esclerose nodular; – com celularidade mista; e – com depleção linfocitária (classificação de Rye).
A esclerose nodular, com um excelente prognóstico, e surgindo em geral sob a forma localizada, predomina em adolescentes e adultos jovens; por sua vez, corresponde ao tipo histológico mais habitual entre nós e nos países desenvolvidos.
A celularidade mista predomina em crianças mais jovens.
A depleção linfocitária surge associada a formas generalizadas da doença, de prognóstico mais reservado.
Após o diagnóstico, é indispensável a caracterização do estádio evolutivo para programar a terapêutica. Usa-se habitualmente a classificação de Ann Arbor, englobando quatro estádios, em função:
- do número e localização dos territórios ganglionares afectados e;
- da eventual infiltração de estruturas não linfóides, como o pulmão, o fígado, ou a medula óssea.
Pormenorizando:
- o estádio I corresponde a compromisso de um único gânglio ou de um só órgão ou local extralinfático; e que;
- os estádios II e III correspondem ao compromisso ganglionar (2 ou mais gânglios), e/ou de órgão ou tecido extraganglionar, respectivamente, dum lado ou dos dois lados do diafragma, o qual serve como referência topográfica.
- o estádio IV corresponde à forma disseminada, com vários órgãos ou tecidos extralinfáticos afectados, com ou sem compromisso ganglionar.
Para determinação do estádio, usam-se várias técnicas imagiológicas como ecografia, tomografia axial computadorizada (incluindo a de alta definição, com emissão de positrões), ressonância magnética e ou estudos isotópicos como as cintigrafias com gálio e tecnécio. Trata-se de técnicas complementares, que adicionam à informação anatómica outros dados sobre a actividade da doença.
Salienta-se que tais técnicas sofisticadas permitem a identificação relativamente rigorosa do estádio, sem necessidade de recurso a técnicas invasivas como a linfangiografia ou a laparotomia exploradora, que pertencem ao passado.
Tratamento
A terapêutica é planeada de acordo com o estádio do doente e a existência ou não, de manifestações associadas a pior prognóstico.
Na idade pediátrica, usam-se esquemas combinados de quimioterapia e radioterapia, com excelentes resultados. Estes esquemas têm vindo a adaptar-se progressivamente, de forma a reduzir a intensidade da quimioterapia e a dose e os campos da radioterapia, sem diminuir a probabilidade de cura. Hoje, é possível obter a cura de doentes usando apenas quimioterapia, com base nos critérios de resposta imagiológica e metabólica aos ciclos inicias de quimioterapia.
Nos casos em que tal resposta não é completa, a terapêutica é complementada com radioterapia, usando doses que não ultrapassam os 20 – 25 Gy, por oposição aos 35 – 40 Gy usados anteriormente. (Nota: 1 unidade de radiação (1 GYY) = 100 rads)
A probabilidade de cura é grande (80 a 90%), mesmo para os estádios mais avançados, com as modalidades modernas de tratamento: duas a seis faixas de quimioterapia, consoante a extensão da doença, seguidas, ou não, de radioterapia.
Os efeitos secundários da quimioterapia são os referidos no capítulo sobre tratamento; considerando-se mais temíveis os efeitos traduzidos pelo aparecimento de neoplasias secundárias, principalmente cancro da tiroideia e da mama, importa, contudo, referir que a sua incidência no contexto de linfoma de Hodgkin é superior à verificada em doentes com outras neoplasias. Este facto parece ser devido ao surgimento e persistência de imunodeficiência celular após cura deste tipo de linfoma.
A este efeito somam-se os efeitos secundários da radioterapia: atrofia das partes moles; perturbações do crescimento ósseo; disfunção da tiroideia; e transformação neoplásica dos tecidos irradiados. Tal significa que estes doentes devem ser seguidos cuidadosamente durante muitos anos numa perspectiva de detecção atempada dos problemas surgidos.
BIBLIOGRAFIA
Abramson SJ, Price AP. Imaging of pediatric lymphomas. Radiol Clin North Am 2008; 46: 313 – 338
Cader FZ, Kearns P, Young L, et al. The contribution of yhe Epstein-Barr virus to the pathogenesis of childhood lymphomas. Cancer Treat Rev 2010; 36: 348 – 353
Castellino SM, Geiger AM, Mertens AC, et al. Morbidity and mortality in long-term survivors of Hodgkin lymphomas: a report from the Childhood Cancer Survivor Study. Blood 2011; 117: 1806 – 1816
Choeyprasert W, Anurathapan U, Pakakasama S, et al. Pediatric non‐Hodgkin lymphoma: Characteristics, stratification, and treatment at a single institute in Thailand. Pediatr Intern 2019; 61: 49-57
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Hoffman R, Benz EJ, Silberstein LE, et al (eds). Hematology: Basic Principles and Practice. Philadelphia: Elsevier, 2018
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Kowsky PL. Manual of Pediatric Hematology and Oncology. Amsterdam: Academic Press, 2005
Madero L, Muñoz A (eds). Hematologia y Oncologia Pediátricas. Madrid: Ergon, 2005
Moro M, Málaga S, Madero L. Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Pizzo PA, Poplack DG (eds). Principals and Practice of Pediatric Oncology. Philadelphia: Lippincott Williams & Wilkins, 2011
Seelisch J, Sung L, Kelly MJ, et al. Identifying clinical practice guidelines for the supportive care of children with cancer: A report from the Children’s Oncology Group. Pediatr Blood & Cancer 2019;66: e27471
Sierrasesumaga L, Antillon F (eds). Tratado de Oncologia Pediátrica. Madrid: Pearson, 2005
Weinstein HJ, Husdon MM, Link MP. Pediatric Lymphomas. Berlin: Springer, 2007
Weinstock MS, Patel NA, Smith LP. Pediatric cervical lymphadenopathy. Pediatr Rev 2018; 39: 433-443