Introdução aos linfomas
Os linfomas na sua generalidade, representando cerca de 15% do total de tumores na idade pediátrica, constituem o terceiro grupo de doenças neoplásicas mais frequentes, depois das leucemias e dos tumores do SNC.
A incidência aumenta ao longo da vida, salientando-se a sobrevivência de > 80% nos países industrializados. Pouco frequentes em menores de 4 anos, alcançam uma incidência anual ~5,7 casos por milhão de menores de 15 anos, e de ~ 34,7 casos por milhão de adolescentes entre 15 e 18 anos.
Dividem-se em dois grandes grupos: Linfomas não Hodgkin e Linfomas de Hodgkin.
Neste capítulo são abordados os linfomas não Hodgkin.
Definição e aspectos epidemiológicos
Os linfomas não Hodgkin são neoplasias de linfócitos maduros ou de células precursoras dos linfócitos que, por mutação genética, perderam as capacidades de maturação e de apoptose, ou seja, de autodestruição. Ao contrário dos linfomas não Hodgkin do adulto, são de grande agressividade.
Os linfomas não Hodgkin são muito menos frequentes na criança do que no adulto, aumentando a incidência de forma progressiva, com a idade. Podem encontrar-se, no entanto, em crianças muito jovens, por vezes lactentes.
Classificação
A caracterização imunológica dos linfócitos patológicos veio originar uma classificação simples dos linfomas não Hodgkin da criança, o que tem vindo a permitir abandonar progressivamente as inúmeras classificações clássicas, baseadas na morfologia e nas características citoquímicas, pouco claras e em regra sem grande relação com a clínica.
De forma resumida, os linfomas não Hodgkin pediátricos classificam-se actualmente, de acordo com a linhagem linfóide afectada, em linfomas B de linfócitos maduros, linfomas T de linfócitos maduros, e linfomas pré T ou pré B que, como o nome indica, são linfomas de células precursoras não maduras de linhagem T ou B.
Aos linfomas B de células maduras correspondem, na classificação da Organização Mundial de Saúde, uma das classificações antigas mais usadas, as categorias histológicas de linfoma de Burkitt, Burkitt like e linfoma B de grandes células.
Aos linfomas T de células maduras corresponde a categoria histológica de linfoma anaplásico de grandes células na mesma classificação; e aos linfomas pré T ou pré B, a categoria de linfoma linfoblástico.
Como sucede com outras neoplasias, são descritas algumas situações predisponentes de linfomas, em geral relacionadas com imunodeficiência congénita ou adquirida. No entanto, para a maioria dos casos diagnosticados, não se consegue encontrar uma causa, como sucede para a generalidade das neoplasias da criança.
1. LINFOMAS B: Linfoma de Burkitt, Linfoma Burkitt like e Linfoma B de grandes células
Definição
O linfoma de Burkitt é, como se referiu, uma neoplasia de linfócitos B maduros que se apresenta morfologicamente como um linfoma de pequenas células redondas, não clivadas.
É muito provavelmente o tumor pediátrico com multiplicação celular mais rápida e crescimento mais veloz.
Formas clínicas
A sua forma endémica foi a primeira a ser descrita, na década de 50, na África equatorial, pelo cirurgião irlandês Burkitt, de quem recebeu o nome. É o tumor mais frequente naquela região de África. Caracteriza-se pela localização preferencial no maxilar superior, podendo atingir igualmente o abdómen e o SNC. Mais tarde, relacionou-se este tumor com o vírus de Epstein-Barr, cujo genoma se encontra quase constantemente no núcleo do linfócito B neoplásico e, também, com a malária, já que a área endémica desta doença é também a área endémica do linfoma de Burkitt.
Admite-se que a infecção pelo plasmódio facilite, pela imunossupressão que lhe é inerente, a proliferação incontrolada dos linfócitos B infectados pelo vírus de Epstein-Barr, causando a doença.
Na União Europeia e nos EUA, esta neoplasia é muito menos frequente, sendo a forma típica de apresentação clínica a de um tumor abdominal de crescimento muito rápido, localizado de início na fossa ilíaca direita e estendendo-se rapidamente a todo o abdómen, o qual se apresenta muito distendido e doloroso. À palpação, encontram-se várias formações tumorais de consistência dura. A ecografia ou a TAC revelam várias massas tumorais intrabdominais, por vezes com infiltração nodular do fígado, baço, ou rins, e adenomegalias mesentéricas e retroperitoneais. Pode haver ascite e derrame pleural.
Com menos frequência, a massa linfóide tumoral, que se localiza de início, preferencialmente, na região terminal do íleo, pode originar um íleo mecânico, que é então a manifestação clínica inaugural. Neste caso, é a cirurgia para resolução do íleo que permite o diagnóstico.
Em estádios avançados o linfoma pode atingir o SNC, com massas tumorais que se localizam principalmente no espaço epidural, e também na medula óssea (comportando-se então como LLA de linfócitos B maduros). Menos frequentemente, o linfoma de Burkitt pode surgir com outras localizações: mediastino; gânglios linfáticos cervicais; e anel de Waldeyer.
Diagnóstico
O diagnóstico é feito por estudos morfológicos, citoquímicos, imunológicos e genéticos. Os dois primeiros revelam a existência de um tumor de pequenas células redondas, não clivadas; os métodos imunológicos permitem a detecção de marcadores de maturidade do linfócito B; e a genética revela as translocações típicas: t (8;14), t(2;8) e t(8;22). O material para estes estudos pode obter-se por citologia do tumor por agulha fina, ou por estudo das células existentes em suspensão no líquido ascítico ou no derrame pleural.
Tratamento
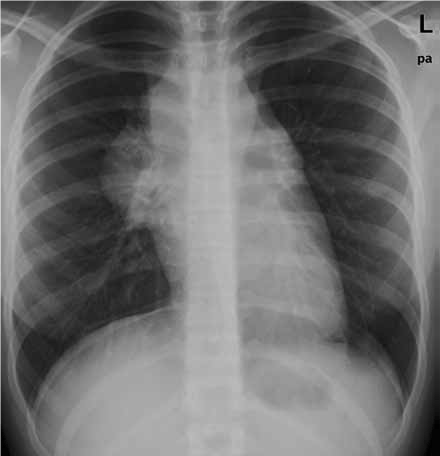
FIGURA 1. Linfoma B difuso de grandes células; radiografia do tórax:adenomegalia mediastínica
O tratamento do linfoma de Burkitt/LLA B constitui um dos maiores sucessos da oncologia moderna. Actualmente, com os modernos protocolos de quimioterapia intensiva, a probabilidade de cura é superior a 90%.
É importante, para além do diagnóstico, caracterizar o estádio da doença, já que os protocolos de quimioterapia possuem vários ramos de intensidade crescente.
Os estádios intermédios tratam-se durante cerca de 4 meses, e as formas mais graves, em que há invasão da medula óssea ou do SNC, durante cerca de 8 meses. Os resultados finais acabam por ser semelhantes.
O linfoma B de grandes células e o linfoma Burkitt like são variantes histológicas na classificação da Organização Mundial de Saúde, mas são igualmente neoplasias de linfócitos B maduros.
Surgem em crianças de grupo etário superior e caracterizam-se pelo aparecimento, não de grandes massas tumorais, como no linfoma de Burkitt, mas de gânglios linfáticos aumentados (adenomegálias) em territórios periféricos ou profundos (intrabdominais e/ou torácicos).
A localização mediastínica (Figura 1) é mais frequente na forma de linfoma B de grandes células do que no linfoma de Burkitt. Este último é tipicamente abdominal, como foi dito.
Embora haja diferenças morfológicas entre estes linfomas B e o linfoma de Burkitt, o tratamento é semelhante e os resultados são igualmente bons.
2. LINFOMAS PRÉ T e PRÉ B: Linfoma linfoblástico
O linfoma pré T, constituído por linfoblastos precursores de linhagem T, é tipicamente supra diafragmático, atingindo o mediastino numa grande percentagem de casos, e também os gânglios dos territórios cervicais, supra claviculares e axilares. Dor torácica, dispneia e disfagia por compressão das vias aéreas ou do esófago, edema e estase venosa do pescoço e parte superior do tórax por compressão da veia cava superior – síndroma da veia cava são as manifestações mais frequentes. (Figura 1)
Em estádios mais avançados, pode haver invasão do SNC ou da medula óssea. Esta última põe problemas de diagnóstico diferencial entre linfoma e leucemia. Por convenção será linfoma se o número de linfoblastos na medula óssea for inferior a 30%.
O linfoma pré B, constituído por linfoblastos precursores de linhagem B, atinge igualmente os territórios linfáticos periféricos, ou profundos, toracoabdominais, não havendo aqui, contudo, volumosas massas tumorais, como sucede no linfoma de Burkitt. Pode, igualmente, em fases avançadas, atingir o SNC ou a medula óssea, pondo iguais problemas de diagnóstico diferencial com leucemia.
A distinção entre leucemia e linfoma é importante já que existem diferenças nos protocolos de tratamento em relação à quimioterapia e à radioterapia.
3. LINFOMA T: Linfoma anaplásico de grandes células
Trata-se de uma entidade nova, durante muito tempo confundida com o linfoma de Hodgkin, hoje diferenciada pelas características imunológicas e genéticas da célula neoplásica.
Manifesta-se por adenomegálias nos territórios periféricos ou toracoabdominais sem, contudo, haver formação de grandes massas tumorais, ao contrário do que sucede com o linfoma de Burkitt, ou o linfoma linfoblástico (o primeiro no abdómen, o segundo no tórax). Pode infiltrar certos órgãos como a pele, o pulmão ou o osso, mas raramente atinge o SNC ou a medula óssea. É, sobretudo, a evolução lenta, com períodos de regressão espontânea, e a repercussão no estado geral, com febre e emagrecimento, que diferenciam este linfoma dos outros e o aproximam do linfoma de Hodgkin.
Não há unanimidade na terapêutica ideal, mas têm sido referidos bons resultados com quimioterapia muito semelhante à utilizada para o linfoma de Burkitt.
BIBLIOGRAFIA
Abramson SJ, Price AP. Imaging of pediatric lymphomas. Radiol Clin North Am 2008; 46: 313 – 338
Cader FZ, Kearns P, Young L, et al. The contribution of yhe Epstein-Barr virus to the pathogenesis of childhood lymphomas. Cancer Treat Rev 2010; 36: 348 – 353
Choeyprasert W, Anurathapan U, Pakakasama S, et al. Pediatric non‐Hodgkin lymphoma: Characteristics, stratification, and treatment at a single institute in Thailand. Pediatr Intern 2019; 61: 49-57
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Hoffman R, Benz EJ, Silberstein LE, et al (eds). Hematology: Basic Principles and Practice. Philadelphia: Elsevier, 2018
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Kowsky PL. Manual of Pediatric Hematology and Oncology. Amsterdam: Academic Press, 2005
Madero L, Muñoz A (eds). Hematologia y Oncologia Pediátricas. Madrid: Ergon, 2005
Moro M, Málaga S, Madero L. Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Pizzo PA, Poplack DG (eds). Principals and Practice of Pediatric Oncology. Philadelphia: Lippincott Williams & Wilkins, 2011
Seelisch J, Sung L, Kelly MJ, et al. Identifying clinical practice guidelines for the supportive care of children with cancer: A report from the Children’s Oncology Group. Pediatr Blood & Cancer 2019;66: e27471
Sierrasesumaga L, Antillon F (eds). Tratado de Oncologia Pediátrica. Madrid: Pearson, 2005
Weinstein HJ, Husdon MM, Link MP. Pediatric Lymphomas. Berlin: Springer, 2007
Weinstock MS, Patel NA, Smith LP. Pediatric cervical lymphadenopathy. Pediatr Rev 2018; 39: 433-443