Introdução
Os tumores neuroblásticos (neuroblastoma, ganglioneuroblastoam e ganglioneuroma) derivam das células pluripotenciais da crista neural que emigram no período embrionário para formar os gânglios simpáticos e a medula suprarrenal. Daí, a sua grande diversidade quanto a localizações anatómicas e a manifestações clínicas.
Definição e aspectos epidemiológicos
O neuroblastoma é, pois, uma neoplasia de origem embrionária com ponto de partida nas células da crista neural. Com uma prevalência de 1/7.000 nados-vivos, constitui o tumor sólido mais frequente no lactente, e o segundo na primeira década da vida, logo depois dos tumores do SNC. Representa cerca de 10% dos tumores malignos na idade pediátrica.
O uso sistemático da ecografia durante a gravidez e no período pós-natal tem permitido o diagnóstico pré-natal e pós-natal imediato dum número crescente de neuroblastomas.
Etiopatogénese
A etiopatogénese do neuroblastoma é desconhecida. De referir a ocorrência de mutações no gene PHOX2B (regulador do desenvolvimento neuronal autonómico), em casos de associação deste tipo de tumores a situações caracterizadas por alterações do desenvolvimento da crista neural (neurocristopatias), como neurofibromatose do tipo 1, síndroma de hipoventilação central e doença de Hirschprung, as quais poderão ser consideradas factores predisponentes.
Por outro lado, mutações genéticas no cromossoma 6p22 estão associadas a risco aumentado de neuroblastoma.
Reportando-nos à teoria de Knudson (ver atrás), cabe referir que a maioria dos casos familiares/hereditários se relaciona com mutações no gene ALK.
A diversidade do comportamento evolutivo do neuroblastoma (adiante salientada) poderá ser explicada pela expressão dos receptores de neurotrofina, determinada geneticamente.
Os grupos de investigação sobre o neuroblastoma consideram importante para o prognóstico – adiante abordado – valorizar a eventual amplificação do oncogene N-myc.
Manifestações clínicas
O espectro clínico é muito variável, existindo muitas interrogações pelo seu comportamento, por vezes, enigmático.
Na verdade, enquanto alguns neuroblastomas metastizados, com repercussão grave inicial sobre o estado geral do doente, podem regredir, espontaneamente ou tratados com quimioterapia de curta duração, pelo contrário, outros neuroblastomas, em estádios mais localizados e sem aparente repercussão geral podem progredir inexoravelmente, apesar dos tratamentos efectuados.
E, quando se fala em evolução do neuroblastoma, importa ainda salientar a evolução possível, quer para ganglioneuroma, benigno, quer para doença com metástases.
De acordo com a sua origem embrionária, o tumor primitivo localiza-se ao longo das estruturas nervosas simpáticas; mais frequentemente no abdómen, na glândula suprarrenal ou ao longo da goteira paravertebral; outras vezes, no tórax, mediastino posterior; mais raramente, na região cervical ou pélvica.
No abdómen, o tumor pode atingir dimensões apreciáveis antes de originar sinais e sintomas. Os mais frequentes são a dor e a distensão abdominal que se manifestam numa criança em regra emagrecida e com aspecto de doença grave, já que, geralmente, a doença se apresenta num estádio avançado.
No tórax, as manifestações são respiratórias, circulatórias ou neurológicas (síndroma de Claude Bernard Horner, por exemplo), devido à compressão das estruturas anatómicas pelo tumor. Nalguns casos, porém, poderá ser um achado ocasional numa radiografia do tórax.
Na região cervical, uma massa tumoral, associada ou não a dor, é o sinal mais frequente.
Como resultado da localização na pelve, surgem alterações do trânsito intestinal ou queixas urinárias, resultantes da compressão do recto ou da bexiga.
Mas, independentemente das localizações anatómicas anteriormente referidas, tratando-se de um tumor paravertebral, pode sempre manifestar-se inicialmente por sinais neurológicos de gravidade variável, quer por compressão das raízes nervosas, quer por invasão do canal medular. Neste último caso, o tratamento torna-se verdadeiramente urgente a fim de evitar sequelas neurológicas graves.
Para além destas manifestações relacionadas com o tumor primitivo, outras podem surgir resultantes da metastização tumoral: dor óssea, por invasão óssea; anemia e trombocitopenia, por invasão da medula óssea; nódulos cutâneos, por invasão da pele; proptose e equimoses palpebrais, por infiltração da órbita; e, em fases muito avançadas da doença, pode assistir-se a metastização pulmonar ou no SNC.
Uma forma peculiar de apresentação que merece referência é a síndroma de Pepper: surge no lactente e caracteriza-se por um tumor localizado na suprarrenal havendo, simultaneamente, infiltração maciça do fígado. A hepatomegália resultante é então a primeira manifestação da doença, observando-se um lactente com estado geral em regra bom, com abdómen volumoso em que se palpa o fígado aumentado de volume.
Algumas vezes esta hepatomegalia é de tal forma exuberante, que se instala um quadro de dificuldade respiratória, e/ou edema do escroto e membros inferiores, e/ou vómitos frequentes e má-nutrição, explicáveis por compressão exercida pelo fígado aumentado de volume sobre as estruturas vizinhas.
Em cerca de 1 a 3% dos doentes podem fazer parte das manifestações clínicas 3 tipos de síndromas paraneoplásicas por:
- VIP (Vasoactive intestinal peptide) – por secreção tumoral de VIP [diarreia secretora intratável, hipopotassémia, distensão abdominal, desidratação, etc.. Está em geral associada a ganglioneuroma ou ganglioneuroblastoma];
- OMA (opsomioclonus-mioclonus-ataxia) – associada a autoanticorpos antineuronais [abalos mioclónicos erráticos, movimentos oculares descontrolados e “olhos e pés dançarinos”, por vezes, ataxia cerebelosa];
- SEC (secreção excessiva de catecolaminas) [taquicárdia, HTA, palpitações, sudação profusa, crises de rubor, sintomatologia que é clássica no feocromocitoma].
Enquanto a patogénese da HTA, no caso de neuroblastoma, tem forte componente de causa renovascular, no feocromocitoma é, sobretudo, comparticipada pelas catecolaminas com um abdómen muito volumoso, em que se palpa o fígado aumentado de volume.
Sob o ponto de vista semiológico, o neuroblastoma apresenta-se frequentemente como uma massa abdominal de consistência endurecida, com superfície lisa, lobulada, fixa, ultrapassando a linha média, ocupando a loca renal e deslocando o rim para baixo e para fora. O diagnóstico diferencial pode fazer-se com o tumor de Wilms, salientando-se que, nesta última situação, a massa não ultrapassa a linha média, sendo em cerca de 5% dos casos bilateral.
Para avaliar o estádio ou grau de extensão da doença (“estadiamento”), diversos grupos cooperativos de investigação a nível mundial consideram fundamentalmente dois critérios: – o International Neuroblastoma Staging System relativo ao estadiamento pós-cirúrgico (estádios 1, 2A, 2B, 3, 4, 4S); e – o International Neuroblastoma Risk Group Staging System relativo ao estadiamento pré-cirúrgico (estádios L1, L2, M, MS).
Para avaliar a resposta ao tratamento consideram-se os critérios internacionais designados por International Neuroblastoma Response Criteria considerando as respostas adiante definidas pelas siglas RC, MBRP, RP, RM, EE, PE.
Dadas as características generalistas desta obra e considerando que os critérios em pormenor podem ser consultados através da bibliografia, apenas se resumem os estádios 1, L1, MS, e RC:
- 1: Tumor localizado com excisão macroscópica completa, com doença residual microscópica ou sem ela; gânglios linfáticos ipsilaterais microscopicamente negativos para o tumor.
- L1: Tumor localizado não afectando estruturas vitais .
- MS: Doença metastática em menores de 18 meses com metástases confinadas à pele, fígado e ou medula óssea.
- RC: Resposta completa: desaparição total do tumor, sem indícios de doença; concentrações normais de ácido vanilmandélico/VMA e de ácido homovanílico/HVA.
O Quadro 1 resume as manifestações clínicas mais e menos frequentes do neuroblastoma. A Figura 1 mostra um lactente com distensão abdominal e hepatomegália.
QUADRO 1 – Clínica do Neuroblastoma
| Manifestações frequentes |
|
| Manifestações raras |
|
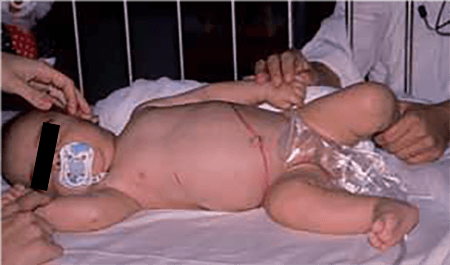
FIGURA 1. Lactente com distensão abdominal por hepatomegália relacionada com neuroblastoma (NIHDE).
Exames complementares e diagnóstico diferencial
Dada a diversidade de manifestações clínicas do neuroblastoma, tal situação poderá ser confundida com outras neoplasias ou situações não neoplásicas. As dificuldades poderão ser maiores nos casos de neuroblastomas que não produzem catecolaminas em excesso, e em cerca de 1% dos casos em que não é obvia a identificação de tumor primário.
As síndromas VIP podem ser confundidas com doença inflamatória intestinal, e os casos com manifestações de OMA poderão corresponder a uma situação neurológica primária.
As equimoses periorbitárias poderão levantar a hipótese de maus tratos/abuso, pelo que a anamnese se torna fundamental. A verificação de anemia, neutropénia, ou trombocitopénia pode levar, em função do contexto clínico ao diagnóstico diferencial com leucemia.
O estudo imagiológico do doente por TAC ou TAC com emissão de positrões (PET) e ou ressonância magnética (RM) revela um tumor de localização e dimensões variáveis, muitas vezes com calcificações, as quais são sugestivas do diagnóstico.
As Figuras 2 e 3 exibem imagens de neuroblastoma de localização intratorácia.
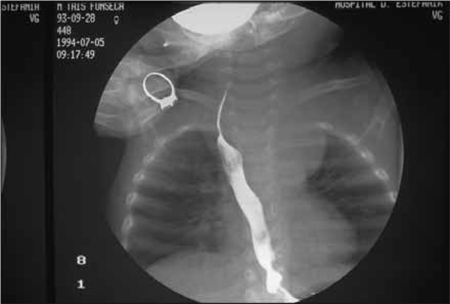
FIGURA 2. Imagem opaca arredondada paravertebral torácica superior de neuroblastoma desviando o esófago visualizado com contraste. (NIHDE)
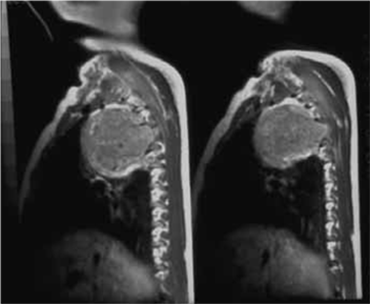
FIGURA 3. Imagem de TAC torácica de perfil evidenciando tumor esférico pré-vertebral (neuroblastoma) ocupando praticamente o terço superior da cavidade torácica. (NIHDE)
Na Figura 2 (radiografia de tórax), em incidência póstero-anterior, observa-se opacidade para vertebral de contorno arredondado ao nível de D1-D4 desviando o esófago contrastado.
A Figura 3 mostra a imagem de um neuroblastoma de localização pré-vertebral superior intratorácia (D2-D7) de contorno arredondado e grandes dimensões (TAC de perfil).
O estudo isotópico com injecção de metaiodobenzilguanidina (MIBG), metabólito que é fixado electivamente pelas células do neuroblastoma, permite determinar com precisão a localização do tumor primitivo e suas metástases, tendo também importância no seguimento dos doentes.
Tratando-se de um tumor produtor de catecolaminas (em 90% dos casos), estas podem ser doseadas na urina, encontrando-se, em geral, aumentadas no início da doença, normalizando com o tratamento. Os ácidos vanilmandélico (VMA) e homovanílico (HVA) são assim importantes, não só no diagnóstico, mas também no estudo evolutivo.
[Nota: valores de referência: VMA (urina): 83±26µg/kg/dia (ou 2-12µg/mg de creatinina); HVA (urina): 3-16µg/mg de creatinina; catecolaminas totais (urina): 0,4-2µg/kg/dia].O mesmo se aplica à enolase sérica e LDH, marcadores que, não sendo específicos, se encontram elevados nas formas mais avançadas da doença.
O diagnóstico é confirmado por exame citológico ou histológico do tumor, obtido por citologia por agulha fina ou por biópsia, colhendo-se igualmente material para estudos genéticos (para classificação genómica).
Actualmente, utilizam-se marcadores genéticos com valor prognóstico, tais como:
- deleção do cromossoma 1, amplificação e mutações no cromossoma 6p22 ;
- amplificação do oncogene N myc.
Em síntese: os requisitos mínimos para estabelecer o diagnóstico de neuroblastoma são:
- diagnóstico anatomopatológico do tecido tumoral com imuno-histoquímica ou sem ela, microscopia electrónica ou elevação do nível de catecolaminas urinárias;
- infiltração da medula óssea (aspirado ou biópsia) por células tumorais e aumento da excreção urinária de catecolaminas.
Tratamento
O neuroblastoma é um tumor quimio e radiossensível. O tratamento é programado de acordo com os critérios que definem o prognóstico do doente no início. Entre estes destacam-se: a idade (inferior ou superior a um ano); o estádio do tumor (localizado e ressecável, localizado e irressecável, disseminado); os marcadores genéticos (N-mys, CHD5 ); e o tipo histológico (favorável, desfavorável).
Assim, alguns doentes serão apenas sujeitos a cirurgia; outros serão submetidos a quimioterapia e cirurgia; outros, ainda, após quimioterapia e cirurgia serão sujeitos a megaterapia com auto transplantação com células estaminais, complementada posteriormente com radioterapia sobre o leito tumoral.
Alguns protocolos prevêem ainda terapia sobre a doença residual persistente após os tratamentos anteriormente referidos, a qual se ensaia com o uso de anticorpos monoclonais, ou com indutores da maturação do neuroblasto (como a isotretinoína), ou ainda com terapia com radioisótopos.
Prognóstico
A probabilidade de cura depende dos vários factores prognósticos acima descritos:
- muito elevada nos estádios localizados sem marcadores genéticos de mau prognóstico e, ao invés,
- reduzida nos estádios avançados ou com marcadores genéticos desfavoráveis, apesar das terapêuticas intensivas a que estes últimos doentes são actualmente sujeitos.
Como marcadores citogenéticos de prognóstico mais reservado de neuroblastoma citam-se:
- a amplificação do proto-oncogene MYCN (ou N-myc) e
- a identificação do gene supressor CHD5 relacionado com deleção 1p36.1.
Relacionando tais noções com a classificação por estadiamento anteriormente descrita, importa destacar alguns dados decorrentes de estudos realizados:
- a amplificação do oncogene N-myc localizado em 2p24 é evidenciada em 20% dos neuroblatomas, sobretudo em formas avançadas (33%) e, em menor proporção, nos estádios 1, 2, 3 e 4S ;
- nos pacientes com neuroblastoma no estádio L1 de qualquer idade e no estádio MS, todos eles sem amplificação do gene N-myc, o prognóstico é excelente; nestes casos é requerido tratamento mínimo, em geral, apenas cirurgia, exceptuando na eventualidade de sintomatologia de compressão medular ou compromisso hepático.
BIBLIOGRAFIA
Brodeur GM. Spontaneous regression of neuroblastoma. Cell Tissue Res 2108; 372: 277-286
Cheung NK, Zhang J, Lu C, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012; 307: 1062 – 1071
Elzembely MM, Dahlberg AE, Pinto N, et al. Late effects in high-risk neuroblastoma survivors treated with high-dose chemotherapy and stem cell rescue. Ped Blood & Cancer 2019; 66: e27421
Granata C, Fagnani AM, Gambini C, et al. Features and outcome of neuroblastoma detected before birth. J Pediatr Surg 2000; 35: 88 – 91
Kliegman RM, Stanton BF, StGeme JW, Schor NF (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2015
Kowsky PL. Manual of Pediatric Hematology and Oncology. Amsterdam: Academic Press, 2005
MacFarland S, Bagatell R. Advances in neuroblastoma therapy. Curr Opin Pediatr 2019; 31:14-20
Madero L, Muñoz A (eds). Hematologia y Oncologia Pediátricas. Madrid: Ergon, 2005
Matthay KK, Shulkin B, Ladenstein R, et al. Criteria for evaluation of disease extent by (123)I-metaiodobenzylguanidine scans in neuroblastoma: a report for the International Neroblastoma Risk Group (INRG) Task Force. Br J Cancer 2010; 102: 1319 – 1326
Moro M, Málaga S, Madero L. Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Pizzo PA, Poplack DG (eds). Principals and Practice of Pediatric Oncology. Philadelphia: Lippincott Williams & Wilkins, 2011
Rubie H, De Bernardi B, Gerrard M, et al. Excellent outcome with reduced treatment in infants with non metastatic and unresectable neuroblastoma without MYCN amplification. J Clin Oncol 2011; 29: 449 – 455
Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA (eds). Rudolph´s Pediatrics. New York: McGraw – Hill Medical, 2011
Sharma R, Mer J, Lion A, Vik TA. Clinical presentation, evaluation, and management of neuroblastoma. Pediatr Rev 2018; 39: 194-203; DOI: 10.1542/pir.2017-0087
Sierrasesumaga L, Antillon F (eds). Tratado de Oncologia Pediátrica. Madrid: Pearson, 2005
Stefan DC, Rodriguez-Galindo C (eds). Pediatric Hematology-Oncology in Countries With Limited Resources. Berlin: Springer, 2014
Zimmerman MW, Liu Y, He S, et al. MYC drives a subset of high risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov 2018; 8: 320-335