Definição
A doença de Hirschsprung (DH) ou megacólon aganglionar congénito é caracterizada pela ausência de células ganglionares na porção mais distal do cólon e recto, característica anatomopatológica que se pode estender proximalmente de modo variável. Tal afecção é uma causa frequente de obstrução intestinal no recém-nascido (RN) e na primeira infância.
A doença de Hirschsprung é classificada como clássica (envolvimento recto-sigmóide: 75% dos casos); longa (envolvimento até ao cólon transverso: 17% dos casos), e extralonga (envolvimento até à válvula íleocecal com possível compromisso do íleo terminal: 8% dos casos). A aganglionose intestinal total, a forma mais grave de doença, é extremamente rara.
A DH surge com uma incidência de cerca de 1/5.000 nascimentos, sendo mais frequente no sexo masculino (4/1). A taxa de incidência familiar oscila entre 4% e 7%.
Etiopatogénese
As células ganglionares entéricas são originárias da crista neural. Estas células estão presentes no intestino anterior à 4ª semana de gestação e iniciam a sua migração na direcção crânio-caudal entre a 5ª e a 12ª semana.
Após a 12ª semana de gestação é iniciada a migração transmural das células para se formarem os plexos mientéricos e os plexos submucosos, processo que termina cerca da 16ª semana.
A causa da ausência de células neurais na parede intestinal deriva de vários factores, como: interrupção da migração crânio-caudal; falência de diferenciação celular após a migração completa devido a alterações da matriz extracelular onde neuroblastos se fixam e se diferenciam; e mecanismo imunogénico mediado pelo “complexo major de histocompatibilidade”, responsável por formação de anticorpos antineuroblastos.
A migração e diferenciação das células da crista neural são reguladas por uma variedade de factores neuropáticos segregados pelas células mesenquimatosas. A expressão destas moléculas, por sua vez, é regulada por vários genes ou vias genéticas (por ex. gene RET e via do receptor da endotelina tipo B).
O padrão de hereditariedade é variável, relacionando-se com o comprimento do intesino envolvido. Quer na DH isolada, quer na associada a síndromas ou outras doenças, foram descritas todas as formas de hereditariedade mendeliana. Até à actualidade identificaram-se 12 genes em diferentes cromossomas cujas mutações podem estar relacionadas com a afecção; como mais importantes consideram-se as do gene RET, localizado no braço longo do cromossoma 10.
Assim, os factores genéticos têm uma importância fulcral na patogénese da doença de Hirschsprung, nomeadamente após a identificação da variação genética responsável pela supressão da expressão celular das células pluripotenciais da crista neural. A doença de Hirschsprung é, pois, englobada no grupo das neurocristopatias, estando intimamente associada a outras doenças ou síndromas que partilham a mesma natureza genética como síndromas de Waardenburg, de Von Recklinghausen, de Smith-Lemi-Opitz, de Jeune, de Joubert, de Down, etc..
Quer na DH isolada, quer na associada a síndromas ou outras doenças, foram descritas todas as formas de hereditariedade mendeliana. Até à actualidade identificaram-se 12 genes em diferentes cromossomas cujas mutações podem estar relacionadas com a afecção; como mais importantes consideram-se as do gene RET, localizado no braço longo do cromossoma 10.
O aspecto fisiopatólogico básico desta doença é a ausência de coordenação celular da actividade motora das fibras colinérgicas pré-ganglionares, e do efeito inibitório das fibras adrenérgicas pós-ganglionares. Assim, desenvolve-se hiperplasia nervosa colinérgica com aumento de produção não inibida de acetilcolina pelos neurónios colinérgicos, e aumento de sensibilidade do músculo liso a esta substância; tal se explica pela ausência de receptores alfa-2 da mediação noradrenérgica, o que impede a contractilidade do segmento agangliónico.
O aspecto patológico macroscópico característico deste problema é a dilatação e hipertrofia do cólon proximal, com abrupta ou gradual transição (cone de transição), para a porção distal, de dimensão normal ou diminuída.
Anatomia patológica
O aspecto histológico é caracterizado por uma ausência de células ganglionares nos plexos mientérico e subcutâneo, e a presença de troncos nervosos não mielinizados hipertrofiados no espaço normalmente ocupado pelas células ganglionares.
Manifestações clínicas e diagnóstico
Em cerca de noventa por cento dos doentes com doença de Hirschsprung o diagnóstico é feito durante o período neonatal. O atraso na emissão de mecónio é o sinal clínico neonatal cardinal desta doença.
Cerca de 90% dos recém-nascidos (RN) com DH eliminam mecónio após as 24 h de vida. Este sinal clínico é seguido por obstipação, vómitos e distensão abdominal nos primeiros dias de vida. O exame rectal revela, classicamente, uma ampola rectal vazia de fezes com uma posterior descarga de fezes líquidas e semi-líquidas de cheiro fétido. A estimulação rectal, o uso de clisteres de limpeza, de laxantes e de emolientes pode fazer regredir o quadro clínico temporariamente. Nesses casos a doença reveste-se de uma forma crónica com períodos de agudização e, manifesta-se clinicamente como um quadro de obstipação crónica com ou sem distensão abdominal apreciável.
Em cerca de um terço destes doentes surge como episódio inaugural um quadro de diarreia aguda profusa. Este sinal clínico é indicativo da possibilidade de desenvolvimento de enterocolite grave que permanece como a principal causa de morte do RN com DH. Nos casos mais graves é caracterizada por distensão súbita, vómito bilioso, febre, sinais de desidratação grave, diarreia sanguinolenta, sépsis e falência multiorgânica.
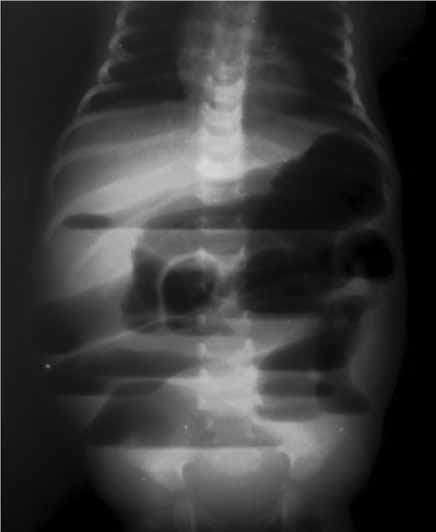
FIGURA 1. Sinais radiológicos de oclusão intestinal no RN no contexto de DH: distensão abdominal e níveis hidroaéreos. (NIHDE)
O diagnóstico da DH depende da conjugação da clínica com o estudo imagiológico, o estudo manométrico e, por fim, o estudo histológico.
Os sinais radiológicos característicos em radiologia convencional são a presença de distensão gasosa de ansas, níveis hidroaéreos e ausência de conteúdo gasoso na região pélvica (cut-off sign) (Figura 1). Nos casos de enterocolite, a distensão gasosa de ansas é muito volumosa, existindo edema da parede, modelagem e irregularidade mucosa identificável. O pneumoperitoneu pode estar presente por necrose transmural e perfuração da parede de ansa.
O clister opaco é um exame radiológico considerado de excelência para o diagnóstico da doença. Este exame permite identificar a zona de espasmo rectosigmóide ou cólico e também a zona de transição (cone de transição) existente entre a zona de espasmo e a zona de dilatação intestinal. A retenção de contraste endoluminal por mais de vinte e quatro horas é muito sugestiva desta patologia, podendo tornar evidente uma zona de transição não imediatamente identificável no início da realização do exame.
A manometria anorrectal (MAR), baseia-se no princípio da ausência de relaxamento do esfíncter interno após a estimulação por aumento de pressão endoluminal pelo balão da sonda. Este fenómeno é característico do segmento aganglionar e, por isso, pode servir como exame de rastreio da doença.
O exame histológico é imprescindível para o diagnóstico definitivo. A biópsia pode ser realizada por meio de acesso laparoscópico (“mapeamento” cólico) ou, mais simplesmente, por meio de biópsia rectal. A biópsia rectal pode ser bem sucedida utilizando uma pinça de sucção, ou por secção cirúrgica. O estudo histológico permite, por análise imuno-histoquímica, identificar a existência, a natureza e maturidade das células ganglionares, assim como a presença de hipertrofia dos troncos nervosos.
Indicação operatória
A DH tem sempre indicação operatória. O princípio geral da terapêutica cirúrgica da DH é a ressecção segmentar do porção recto-sigmóide-cólica aganglionar e o abaixamento do cólon normal gangliónico até à margem do ânus. Até à realização da cirurgia definitiva o RN é mantido num programa de descompressão cólica por meio de clisteres de limpeza denominado classicamente nursing.
Como nova modalidade terapêutica, considerada ainda em fase de investigação, cita-se a transplantação de células-mãe do sistema nervoso entérico no cólon aganglionar.
Complicações pós-operatórias
As complicações pós-operatórios na DH são decorrentes de dois aspectos fundamentais: por um lado, complicações associadas ao abaixamento cólico e da anastomose colo-rectal: infecção local, deiscência e isquémia do segmento cólico mobilizado e estenose da anastomose colo-rectal; por outro, pode surgir uma complicação funcional – as células ganglionares embora presentes, têm uma disposição anómala e displástica condicionando obstrução funcional cólica distal.
A complicação mais grave é a enterocolite pós-operatória. É caracterizada por distensão abdominal extrema, hipertermia, diarreia paradoxal profusa e hemática, e por síndroma séptica. Esta situação obriga a descompressão intestinal de urgência por sonda de enteroclise e instituição de antibioticoterapia de largo espectro e de pausa alimentar. A enterocolite pós-operatória, que pode ocorrer em cerca de 20% dos casos, constitui a primeira causa de morte pós-operatória nestes doentes.
Seguimento
O seguimento dos doentes com DH deve ter em conta, não só a evolução pós-operatória, como também o status funcional intestinal e o desenvolvimento geral da criança.
Numa situação de boa evolução cirúrgica, com um bom funcionamento do segmento cólico mobilizado, é provável a ausência de obstipação grave pós-operatória e boa evolução estaturo-ponderal associada.
Todas as complicações decorrentes do quadro de retenção fecal, estase fecal, proliferação bacteriana e má-absorção, podem ser ultrapassadas com a resolução cirúrgica da doença.
Prognóstico
Na ausência de complicações mecânicas e funcionais, e de episódio ou episódios de enterocolite pós-operatória, o prognóstico final da DH é bom, com resultados de cerca de 90% de cura.
BIBLIOGRAFIA
Amiel J, Sproat E, Garcia-Barceló M, et al. Hirschprung disease, associated syndromes and genetics: a review. J Med Genet 2008; 45:1-14
De Lorijn F, Reitsma JB, Voskuijl WP, et al. Diagnosis of Hirschsprung’s disease: A prospective comparative accuracy study of common tests. J Pediatr 2005; 146: 787-792
Feldman T, Wershil BK. In Brief: Hirschsprung Disease Pediatr Rev 2006; 27: e56-e57; DOI: 10.1542/pir.27-8-e56
Fujimoto T, Puri P. Persistent enterocolitis following diversion fecal stream in HD: a study in mucosal defense mecanisms. Ped Surg Int 1998; 3: 141-114
Fujimoto T, Reen D, Puri P Immunohistochemical characterisation of abnormal innervation of colon in HD. J Ped Surg 1987; 22: 246-249
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Gunnarsdóttir A, Sandblom G. Quality of life in adults operated on for Hirschprung disease in childhood: J Pediatr Gastroenterol Nutr 2010; 51: 160 – 166
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Neill Jr JA, Rowe MI, Grosfeld JL, et al (eds). Pediatric Surgery. Philadelphia: Elsevier, 2017
Skinner M. Hirschsprung’s disease. Curr Prob Surg 1996; 33:389-392
Teitelbaum DH, Coran AG. Reoperative surgery for hirschsprung’s disease. Sem Pediatr Surg 2003; 12: 124-131
Thapar N. New frontiers in the treatment of Hirschprung disease. J Pediatr Gastroenterol Nutr 2009; 48 (Suppl): S92- S94
Willie R, Hyams JS, Kay M (eds). Pediatric Gastroenterology and Liver Disease. Philadelphia: Elsevier, 2016