Definições
Num sentido lato, Anomalias Congénitas (AC) – termo sinónimo de malformações congénitas/defeitos congénitos – são erros de desenvolvimento, presentes desde o período embriofetal e manifestando-se por alterações estruturais, funcionais ou bioquímicas, que podem ser detectadas ao nascer ou mais tardiamente.
A sua etiologia é heterogénea, inerente ao feto como no caso das anomalias cromossómicas ou génicas, ou exterior a ele como no caso de factores físicos, infecciosos, bioquímicos ou outros. Muitas vezes pode haver acumulação de factores como no caso da chamada etiologia multifactorial.
Num conceito mais restrito, o termo refere-se a um defeito estrutural de instalação embriofetal, reconhecido ou não ao nascer, e de etiologia variável.
O termo Dismorfologia diz respeito ao estudo das anomalias de forma do organismo humano, assim como dos respectivos mecanismos causais.
Importância do problema
A ocorrência de AC está documentada desde os tempos mais remotos da Humanidade, em muitos textos da Antiguidade, sendo inúmeras as suas representações na Arte em todas as civilizações.
A explicação das suas causas, bem como o comportamento da sociedade, tem variado naturalmente de acordo com as várias culturas e o momento da História. Mas foram os enormes avanços da Genética Médica alcançados nas últimas décadas, e o reconhecimento de factores nocivos do ambiente como causa de anomalias congénitas, que tornaram possível não só os conhecimentos que hoje temos da sua etiologia e epidemiologia, bem como a utilização de métodos de prevenção cada vez mais eficazes.
Hoje as AC são um problema de Saúde Pública e a sua incidência é tanto mais elevada quanto menor for a idade gestacional considerada. Se no período pré-natal é difícil quantificar a sua importância devido ao elevado número de perdas embrionárias e fetais por AC, elas são relativamente frequentes e preocupantes no período pós-natal, uma vez que 2 a 3 por cento dos recém-nascidos vivos têm uma ou várias AC de gravidade muito variável, o que justifica frequentemente o recurso a hospitalizações.
De acordo com estatísticas hospitalares (www.marchofdimes.com/peristats), cerca de 50% dos casos de AC identificados em RN corresponde a defeitos múltiplos; 10% dos que requerem hospitalização, corresponde a situações do foro genético; em 18% a etiologia é desconhecida; e em 40% é requerida correcção cirúrgica.
No que respeita à comparticipação das AC na mortalidade neonatal, nos USA, em 2015, foram obtidos os seguintes dados: 137 óbitos/100.000 nados-vivos; para comparação, os valores obtidos quanto a mortalidade neonatal por outras causas foram os seguintes: prematuridade e ou baixo peso – 109/100.000; síndroma de morte súbita/SMS/SIDS – 55/100.000.
É clássica a comparação das AC a um iceberg. As que se evidenciam após o nascimento, representadas pela parte visível da massa gelada, são apenas uma pequena parcela da realidade. Na verdade, a maioria das AC, particularmente as mais devastadoras, são letais no período pré-natal:
- Cerca de 40% dos zigotos não sobrevivem devido a erros de desenvolvimento, particularmente durante as primeiras oito semanas;
- 2 a 3% dos recém-nascidos (RN) têm anomalias congénitas, a maioria das quais de natureza genética;
- Das mais de 4.000 doenças mendelianas indexadas no catálogo de doenças hereditárias de McKusick, cerca de 1.900 têm alterações da morfogénese, sendo para cima de 1.000 as descritas com conjuntos malformativos complexos.
Etiopatogénese
Como complemento do que foi referido na alínea anterior, o Quadro 1 resume os factores etiológicos mais frequentemente implicados: genéticos e ambientais (teratogénicos), por vezes associados; pode concluir-se que, na maioria dos casos não é possível identificar o factor causal.
QUADRO 1 – Anomalias congénitas – Etiologia
Jones Kl, 2997 |
| Etiologia |
· Factores de Ambiente (Teratogénicos) (~10%) · Factores genéticos (~10-25%)
· Factores Ambientais e Genéticos · Factores Desconhecidos (~65-75%) |
QUADRO 2 – Anomalias congénitas – Factores ambientais
Jones Kl, 2997 |
| Factores Ambientais (Teratogénicos) |
· Germes Microbianos
|
· Doenças Maternas
|
· Agentes Químicos, Físicos, Drogas
|
No Quadro 2 são referidos alguns exemplos de factores ambientais (teratogénicos).
Desenvolvimento embrio-fetal normal e patológico – Breves conceitos
O genoma que o zigoto recebe dos seus progenitores constitui um conjunto de regras que permite construir um embrião. Essas regras, que constituem o mecanismo regulador do desenvolvimento embrionário, estão na base de uma sucessão muito complexa de acontecimentos minuciosamente programados no tempo e no espaço.
Desses acontecimentos fazem parte processos tão importantes como a divisão celular, a adesão celular, a indução, a migração das células, a apoptose, o crescimento e a diferenciação.
Os genes são os “instrumentos” moleculares responsáveis pela organização de toda a morfogénese e estrutura cromossómica. Convém, no entanto, ter sempre presente que num cariótipo se vêem os cromossomas mas não se visualizam os genes.
Cabe à biologia molecular explicar como a informação unidimensional contida na cadeia de ácido desoxirribonucleico (ADN) origina uma informação tridimensional (proteína) responsável pelas transformações têmporo-espaciais que caracterizam o normal desenvolvimento do embrião.
A partir do ovo, o embrião tem, pois, teoricamente todas as potencialidades para se desenvolver e crescer de uma forma harmoniosa e previsível. Esta evolução está dependente da interacção de factores genéticos específicos de cada indivíduo e de factores ambientais muito diversos com particular relevância para os factores nutricionais, endócrinos e metabólicos.
O programa de crescimento e desenvolvimento do embrião é muito preciso no que respeita ao tempo e ao espaço em que ocorrem os acontecimentos que irão transformar o ovo num recém-nascido.
Com uma frequência muito maior do que seria de esperar e do que seria desejável, existem falhas de natureza genética ou epigenética que conduzem a uma disrupção do programa estabelecido com consequências mais ou menos graves na estrutura e funcionamento do embrião.
É muito útil para compreender a génese das anomalias congénitas, relembrar os fenómenos da fertilização e as fases do desenvolvimento embrio-fetal , caracterizadas por uma sucessão de estádios ininterruptos mas morfologicamente bem definidos.
A fertilização é um fenómeno complexo de interacção entre um óvulo e um espermatozóide, veículos da informação genética materna e paterna, indispensável ao normal desenvolvimento do embrião e do feto. A fertilização tem como consequência a formação do zigoto, considerado como o ponto zero do desenvolvimento embrionário.
Por vezes, a informação que chega ao zigoto, quer por via materna, quer por via paterna, contém erros de natureza génica ou cromossómica, responsáveis pela génese de anomalias congénitas de natureza e gravidade muito variáveis.
Assim, as anomalias cromossómicas de número (devidas a não-disjunção meiótica), as anomalias cromossómicas de estrutura e as mutações génicas, podem chegar ao zigoto por via materna, paterna, ou ambas simultaneamente.
A anomalia cromossómica mais frequente no RN vivo é a trissomia 21, (Figura 1, Capítulo sobre anomalias cromossómicas) que pode revestir a forma de trissomia livre (Figura 1) ou de trissomia por translocação (translocação 21/14 na Figura 2).

FIGURA 1. Trissomia 21 – Cariótipo (forma livre)

FIGURA 2. Trissomia 21 – Cariótipo (translocação: 21/14)
Neste último caso, é necessário provar se a anomalia é herdada de um dos progenitores ou se é uma situação de novo a fim de poder calcular riscos de repetição.
Mas a não-disjunção pode também ser mitótica (pós-zigótica) conduzindo à formação de mosaicos. De igual modo, as mutações génicas podem aparecer só nas primeiras fases do desenvolvimento, com consequências variáveis em termos de expressão fenotípica.
Nas primeiras 24 horas que se seguem à fusão dos pronúcleos feminino e masculino, inicia-se uma série de divisões mitóticas de forma que no 4º dia existe um conjunto de 32 células constituindo a mórula.
Na fase de mórula, cada uma das células que a compõem pode exprimir todo o potencial genético do novo indivíduo e uma só célula pode dar origem a um indivíduo. Estas células pluripotenciais totipotentes, quando confrontadas com erros genéticos ou agressões ambientais, têm uma grande capacidade de se intersubstituir podendo, assim, compensar esses erros. Se não forem capazes de o fazer, o destino do embrião será a morte. Este fenómeno que é conhecido como a lei do tudo ou nada, tem muita importância quando é necessário avaliar o risco de aparecimento de anomalias congénitas em caso de agressão teratogénica nesta fase do desenvolvimento.
A partir do 4º dia de vida a mórula começa a absorver líquido dando lugar à formação de uma cavidade interna; toma então o nome de blastocisto que se vai implantar na parede uterina por volta do 6º dia. No fim da primeira semana o embrião é unilaminar.
Entretanto a capacidade totipotente das células perde-se e, com o blastocisto, começa uma fase de especialização celular. As células tornam-se pluripotentes, isto é, são capazes de se diferenciar em quase todos os tecidos embrionários excluindo a placenta e anexos.
A partir da segunda semana dá-se a formação do embrioblasto, cujo destino é o desenvolvimento do embrião e do trofoblasto originando o desenvolvimento da placenta. No fim da segunda semana o embrião é bilaminar.
Durante a terceira semana forma-se o embrião trilaminar com o disco embrionário tridérmico que dará origem à ectoderme, mesoderme e endoderme e, posteriormente, a todos os tecidos e órgãos definitivos.
Durante a quarta semana do desenvolvimento têm lugar transformacões muito complexas e rápidas que marcam a passagem para a organogénese.
A estas quatro primeiras semanas, em que se dão os acontecimentos mais importantes em termos de desenvolvimento embrionário, dá-se o nome genérico de blastogénese. Embora muitos embriologistas não atribuam muita importância à individualização destas primeiras quatro semanas no contexto da embriogénese, o facto é que o seu conhecimento é indispensável para compreender a génese das anomalias congénitas.
Assim, é nesta fase que se estabelecem os campos de desenvolvimento, os eixos do embrião, a linha média, a lateralidade e a segmentação, que ocorre a neurulação, a cardioangiogénese, a mesonefrogénese e aparecem os esboços dos membros. A placenta, que também inicia a sua formação durante a blastogénese é naturalmente determinante para a sobrevivência do feto (ver adiante).
Os campos de desenvolvimento têm um enorme interesse na compreensão da génese das anomalias congénitas.
Os defeitos mais graves do desenvolvimento estabelecem-se na blastogénese. Os erros ocorridos nesta fase podem naturalmente dar origem à morte do embrião, ou mais tardiamente do feto, mas podem também conduzir ao nascimento de crianças com anomalias congénitas gravíssimas, interessando um ou mais campos de desenvolvimento.
A partir da quinta semana começa a organogénese que decorre entre o 28º e o 56º dias. São outras quatro semanas, durante as quais se vão formar todos os órgãos, organizando-se em aparelhos ou sistemas. Nesta fase cada órgão e cada sistema tem um momento ou período crítico de formação cujo conhecimento volta a ter muita importância na avaliação do risco teratogénico.
Na organogénese distinguem-se dois processos fundamentais: a morfogénese – formação dos órgãos, e a histogénese – diferenciação das células e organização dos tecidos.
No fim da oitava semana termina organogénese, última fase embriogénese.
O período entre as nove semanas e o nascimento, (período fetal) é dominado pelo crescimento e maturação do feto.
A fenogénese, terceira e última parte do desenvolvimento, prolonga-se para além da vida fetal terminando quando se atinge a maturidade sexual.
Nas Figuras 3 e 4 são apresentados alguns exemplos de anomalias congénitas.
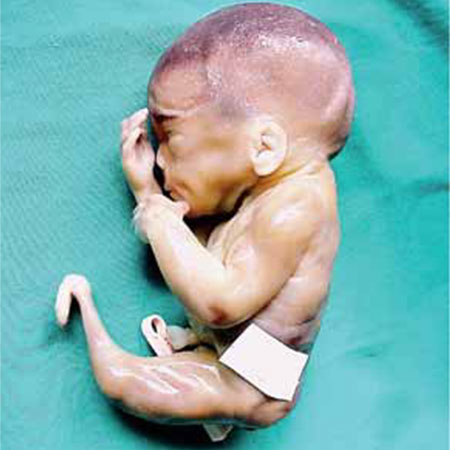
FIGURA 3. Sirenomelia/Embriopatia diabética
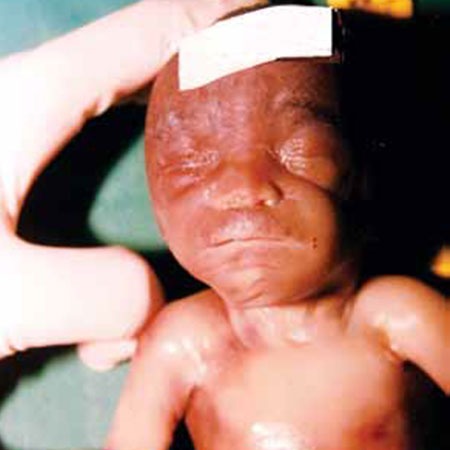
FIGURA 4. Embriofetopatia alcoólica
Figura 3
Feto com 20 semanas de idade gestacional, em que se verifica um único membro inferior constituído por 3 segmentos. O exame radiológico identificou um único fémur alargado e achatado com 2 côndilos, 2 rótulas, 2 tíbias e ossos de pé rudimentares. Havia também imperfuração anal, agenésia renal bilateral e agenésia do útero e restantes estruturas do aparelho genital.
A história revelou diabetes insulinodependente e gravidez seguida de forma irregular.
Trata-se de um defeito da blastogénese.
Diagnóstico – Embriopatia diabética e regressão caudal com sirenomelia.
Figura 4
Feto com 20 semanas de idade gestacional, com cardiopatia congénita. A existência de lábios muito finos num feto de raça negra levou-nos a pôr a hipótese de embriofetopatia alcoólica. A história revelou gravidez não vigiada e mãe com hábitos alcoólicos muito acentuados. Neste caso a valorização de uma anomalia minor foi o fio condutor para o diagnóstico.
O efeito do álcool teve o seu início na embriogénese (cardiopatia) e prolongou-se pela fenogénese com evidência de uma anomalia minor (lábios finos).
Diagnóstico – Embriofetopatia alcoólica.
Campos de desenvolvimento e sua relação com a génese das anomalias congénitas
Na primeira metade do século XX os trabalhos de embriologia experimental de H Spemann e JS Huxley introduziram a noção de campo de desenvolvimento. Em 1982 JM Opitz propunha a sua aplicação em genética clínica e, a partir desse ano, um grupo de trabalho internacional propunha uma nova terminologia para os erros da morfogénese adoptando o conceito de campo de desenvolvimento para explicar a génese da maioria das anomalias congénitas.
Assim, um campo morfogenético ou de desenvolvimento é constituído por uma parte do embrião representando uma unidade coordenada de indução embrionária da qual resulta um conjunto de estruturas anatómicas. Daí decorre que o campo de desenvolvimento é a unidade fundamental do desenvolvimento, também definida como uma unidade reactiva que responde de forma idêntica a agressões diferentes, como anomalias cromossómicas, mutações génicas ou teratogénios.
Na fase inicial da blastogénese a totalidade do embrião constitui um campo de desenvolvimento primário que contém em si próprio o modelo geral do desenvolvimento. Gradualmente, o campo primário divide-se em vários campos progenitores, que são os primórdios das estruturas definitivas.
Os campos progenitores, por sua vez, dão origem aos campos secundários que, já durante a organogénese, serão os responsáveis pelas estruturas finais e irreversíveis do embrião.
Todo este processo aparece, pois, como um conjunto de acontecimentos em cascata e as anomalias serão tanto mais graves e diversificadas quanto mais precoce for o momento em que o erro acontece. Nesta perspectiva, os erros ocorridos na blastogénese durante o estabelecimento dos campos progenitores, devido à sua proximidade e à partilha de mecanismos moleculares, originam anomalias que afectam estruturas diferentes em várias regiões do corpo; são referidas como defeitos politópicos de campo, isto é, envolvem dois ou mais campos progenitores.
As anomalias da blastogénese são heterogéneas do ponto de vista etiológico, graves e altamente letais, com baixo risco de recorrência e afectando predominantemente as estruturas da linha média. Um mesmo conjunto malformativo pode ter etiologias diversas, uma vez que o campo de desenvolvimento reage da mesma maneira a agressões diferentes.
Uma excelente revisão de J. Opitz refere uma extensa lista de anomalias a incluir como defeitos da blastogénese, em que sobressaem a gemelaridade monozigótica, os defeitos politópicos de campo, as associações, as anomalias aparentemente monotópicas mas com provável origem na blastogénese e as anomalias da formação do cordão umbilical e da placenta.
Por outro lado, os erros ocorridos durante a organogénese nos campos secundários originam anomalias limitadas a uma só estrutura ou região do corpo, sendo referidos como defeitos monotópicos de campo. São exemplos as anomalias localizadas tais como fenda palatina, hipospádia ou polidactilia. Mesmo assim, embora se venham a manifestar durante o período da organogénese, a sua origem real pode ter sido durante a blastogénese.
Findo o período da embriogénese, correspondente às oito primeiras semanas de vida do embrião, as estruturas embrionárias estão formadas de uma forma irreversível e assume-se que já não será possível o desenvolvimento de anomalias estruturais graves (ou major).
Durante a fenogénese é possível o aparecimento de anomalias ligeiras (minor); refere-se que pequenas dismorfias faciais podem tornar-se aparentes apenas em fases mais tardias do desenvolvimento embrionário. As anomalias cromossómicas, que produzem os seus efeitos desde a blastogénese, reunem frequentemente anomalias major e minor, o que significa que a sua acção se prolonga durante a fenogénese. (ver adiante)
O mapa génico das anomalias congénitas
O enorme impacte que as técnicas de biologia molecular tiveram no estudo do genoma humano permitiram a construção de um mapa que identifica e localiza os genes em segmentos cromossómicos específicos.
Dado que se trata de uma ciência sempre em expansão, qualquer livro estará sempre parcialmente desactualizado nesta matéria e a consulta de artigos “on-line” é indispensável para uma actualização permanente. Não cabe no âmbito deste livro uma referência extensa aos genes já identificados, mas pode-se dizer que mais de 50% das doenças que constam da última edição do indispensável livro “Smith’s Recognizable Patterns of Human Malformation” já têm genes identificados.
Do conhecimento cada vez mais completo do funcionamento da embriologia molecular decorrem duas observações importantes que são a heterogeneidade alélica e a heterogeneidade génica de certas anomalias isoladas ou múltiplas.
No primeiro caso, mutações diferentes no mesmo gene são responsáveis por fenótipos diferentes. São exemplos as mutações no gene GLI3 localizado no cromossoma 7, que são responsáveis por doenças tão diferentes como a síndroma de Pallister-Hall, a síndroma de Greig ou certas formas de polidactilia isolada. Também a acondroplasia e o nanismo tanatóforo, situações até há pouco tempo consideradas independentes, dependem de mutações diferentes do mesmo gene localizado no cromossoma 4.
No segundo caso, uma mesma síndroma com quadro clínico em tudo semelhante, pode ser devida a mutações em genes diferentes. Temos como exemplo a síndroma de Bardet-Biedl, na qual já se demonstrou a relação causal com vários genes diferentes localizados nos cromossomas 3, 4, 11, 14, 15, 16 e 20.
Ao contrário do que alguns investigadores supunham, o conhecimento dos genes responsáveis pelas AC não diminuiu, mas aumentou a importância da observação clínica cuidadosa, assim como a responsabilidade do sindromalogista, que deve interpretar e construir um padrão de anomalias que possa conduzir a um diagnóstico. Só através deste será possível determinar qual o gene alvo que queremos encontrar.
Classificação
Para efeitos práticos as AC são divididas em major e minor.
As anomalias ditas major são causa de perturbações funcionais ou estéticas de gravidade variável pelo que requerem cuidados médicos ou cirúrgicos como terapia curativa ou paliativa. As anomalias ditas minor são mais frequentes do que as major, mas a sua presença não levanta problemas de natureza funcional ou estética, pelo que não requerem, em geral, qualquer intervenção terapêutica. No entanto, a sua valorização é importante, pois podem constituir um fio condutor para a procura de outras anomalias mais graves que podem ocorrer em conjunto, como é o caso das anomalias renais detectadas através da existência de anomalias minor dos pavilhões auriculares.
Do ponto de vista qualitativo, é útil dividir as anomalias congénitas em quatro subgrupos:
Malformação – consiste num processo anormal de desenvolvimento de natureza intrínseca responsável por um defeito morfológico de um ou mais órgãos. É o que acontece, por exemplo, como consequência de uma anomalia cromossómica.
Disrupção – depende de um acidente grave causado por factores extrínsecos à estrutura do embrião, normal até dada fase do desenvolvimento; tais factores são em geral desconhecidos. De tal resulta um defeito morfológico de um ou mais órgãos. É o que acontece, por exemplo, como consequência da existência de bandas amnióticas.
Deformação – resulta da acção de forças mecânicas extrínsecas ou intrínsecas ao feto, que modificam a forma, o tamanho ou a posição da totalidade do corpo ou de parte dele (com normalidade prévia, até se verificar a acção de tais forças). É o que acontece, por exemplo, como consequência do oligoâmnio.
Displasia – quando há morfogénese anómala com alteração mais ou menos grave da organização celular de um ou vários tecidos. É o que acontece, por exemplo, nas displasias renais ou nas displasias ósseas.
Por vezes é difícil distinguir estes grupos entre si. Mas essa distinção é indispensável em termos de aconselhamento genético, uma vez que as formas de transmissão são diferentes, e diferente o risco de repetição.
As AC podem ser únicas ou múltiplas. É neste último grupo que existe actualmente alguma confusão no que respeita à definição, nomenclatura e limites da variabilidade fenotípica.
Em 1982 formou-se um Grupo de Trabalho Internacional (IWG) liderado por J Spranger, que se debruçou sobre os erros da morfogénese, a sua definição e terminologia. Posteriormente, no Congresso Internacional de Genética reunido em Berlim, em 1986, o mesmo grupo clarificou e redefiniu esses conceitos, de acordo com o conhecimento da etiologia e patogenia dos conjuntos malformativos.
Do ponto de vista quantitativo são consideradas as anomalias que constam do Quadro 3: hipo e hiperplasia, hipo e hipertrofia, atrofia, agenésia e aplasia.
Estes conceitos têm-se revelado de grande utilidade quando se trata de compreender melhor as AC, calcular riscos de repetição e planear diagnóstico pré-natal em futuras gravidezes.
QUADRO 3 – Alterações quantitativas da morfogénese
| Hipoplasia/Hiperplasia |
| · Hipo ou hiperdesenvolvimento de um tecido, órgão ou organismo em função, respectivamente, do nº. diminuído ou aumentado de células. |
| Hipotrofia/Hipertrofia |
| · Hipo ou hiperdesenvolvimento em função das dimensões diminuídas ou aumentadas das células. |
| Agenésia |
| · Ausência de uma parte do corpo devido a ausência do “primordium” |
| Aplasia |
| · Ausência de uma parte do corpo por não desenvolvimento do “primordium” |
| Atrofia |
| · Diminuição das dimensões e/ou nº das células de órgão (s) ou tecido (s) normalmente desenvolvido (s). |
São descritas quatro formas de conjuntos de anomalias (múltiplas):
Síndroma – define-se como um conjunto de anomalias relacionadas entre si, constituindo uma entidade etiologicamente bem definida (génica, cromossómica, teratogénica), embora a patogenia nem sempre possa ser esclarecida. Daqui decorre que a trissomia 21 e a embriofetopatia alcoólica são exemplos de síndromas, e também que “síndroma de etiologia desconhecida”, frase tantas vezes utilizada, não tem sentido.
Associação – define-se como a ocorrência de um conjunto de anomalias de uma forma mais frequente do que o acaso faria supor, e cuja etiologia e patogenia são desconhecidas. Este grupo poderia também ser designado como defeitos da blastogénese de natureza idiopática. Uma associação é habitualmente designada por acrónimos, como por exemplo a associação VACTERL (Vertebral, Anal, Cardiac, fístula Tráqueo-Esofágica, Renal, Limbs) e a associação CHARGE (Coloboma, Heart, Choanal Atresia, Retardation, Genital, Ears).
Mas a etiologia das associações tende naturalmente a ser esclarecida e quando isso acontece, a associação dá lugar a síndroma. Exemplo disso é o que aconteceu com a já mencionada associação CHARGE depois de recentes investigações demonstrando várias mutações no gene CHDZ localizado em 8q12, responsáveis por grande número de casos da associação CHARGE.
Sequência – define-se como um conjunto de anomalias que tem a sua origem numa única anomalia que constitui o acidente primário e que é responsável por um conjunto de acontecimentos em cascata. A etiologia, conhecida ou não, é heterogénea e os mecanismos patogénicos são, evidentemente, conhecidos. Temos como exemplo o mielomeningocele cuja sequência será: defeito de encerramento do tubo neural – desenvolvimento incompleto dos ossos da coluna vertebral com exteriorização da medula (anomalia de Arnold-Chiari) – hidrocefalia e pés botos.
Defeito politópico de campo – este tipo de defeito já foi referido atrás; as anomalias relacionam-se com alterações de dois ou mais campos progenitores.
As anomalias múltiplas, no seu conjunto, estão intimamente relacionadas com os campos de desenvolvimento e os seus erros.
Avaliação clínica
A avaliação clínica das anomalias únicas ou múltiplas, além do seu interesse académico, tem como objectivo último um diagnóstico que permita esclarecer os pais quanto às causas do seu aparecimento, à história natural da doença, à eficácia de eventuais terapêuticas médicas ou cirúrgicas, às formas de transmissão e riscos de recorrência e à possibilidade de eventual diagnóstico pré-natal numa futura gravidez. Este conjunto de actividades define o chamado aconselhamento genético; e para que ele seja possível, torna-se indispensável uma avaliação clínica pormenorizada e a utilização de meios complementares de diagnóstico adequados.
O protocolo habitualmente utilizado no estudo e diagnóstico das anomalias congénitas não é diferente do habitualmente usado em Pediatria, mas envolve algumas particularidades relacionadas com a necessidade de construir um padrão dismorfológico que seja um fio condutor para o diagnóstico de uma entidade conhecida.
Assim, o protocolo deverá incluir:
- Anamnese pessoal e familiar com representação gráfica da árvore genealógica;
- Observação geral e dos parâmetros de desenvolvimento físico, psicomotor e sensorial;
- Descrição da dismorfologia facial;
- Descrição pormenorizada das anomalias presentes;
- Registo fotográfico da face e das anomalias relevantes.
O estudo clínico orientará para os exames complementares necessários a cada caso, salientando-se:
- Exame citogenético com eventual recurso a citogenética molecular;
- Exame radiológico e outros registos imagiológicos;
- Exames de natureza hematológica, bioquímica, enzimática ou outra;
- Estudo génico orientado pela hipótese diagnóstica proposta para cada caso.
Na observação de uma criança com AC reveste-se de particular importância a apreciação do seu aspecto geral (características faciais, forma do corpo, postura, movimento, linguagem e comportamento), de forma a identificá-la por meio de uma comparação subjectiva com outras cujo diagnóstico é conhecido. Esta impressão global ou Gestalt, que se apoia no facto de as várias impressões isoladas (visuais, auditivas e outras) estarem de tal forma organizadas que são percebidas como um todo e não como fenómenos dissociados, leva-nos a identificar uma pessoa conhecida quando a vemos sem necessidade de analisar as suas várias componentes.
A primeira tarefa do especialista em anomalias da forma do organismo ou dismorfologista é, pois, interpretar uma dada constelação de sinais observados no seu doente de forma a identificar uma síndroma, uma associação ou uma sequência. A parte mais difícil desta tarefa reside no facto de não haver, em geral, sinais patognomónicos, o espectro de anomalias poder ser restrito ou vasto dentro de uma mesma entidade, e várias doenças etiologicamente bem definidas partilharem anomalias comuns. A dismorfologia é uma ciência em evolução permanente.
A indispensável definição de critérios mínimos e de limites quanto a expressão fenotípica de uma determinada entidade nem sempre tem reunido o consenso dos dismorfologistas. A tudo isto acresce a contínua publicação de casos clínicos cuja interpretação também nem sempre é coincidente. Com algum sentido de humor, A Verloes apontava recentemente que os sindromalogistas se podem dividir: nos que separam entidades até aí bem definidas em vários subgrupos a que dão novos nomes (splitters); nos que reunem numa entidade única várias outras doenças até aí consideradas como independentes (lumpers); e nos que mudam certos conjuntos de anomalias de uma síndroma para outra (cutters and pasters).
Num futuro próximo e à medida que se forem identificando os genes responsáveis pela génese das AC estes problemas vão perder a sua importância.
Convém, contudo, não esquecer que, em termos de aconselhamento genético e de diagnóstico pré-natal, o reconhecimento clínico de uma entidade e o conhecimento da sua história natural terá sempre importância. Mutações diferentes no mesmo gene podem corresponder a situações clínicas de gravidade muito variável; e, se a variação intrafamiliar não é significativa, não é a presença de uma determinada mutação génica, mas sim o quadro clínico esperado, que poderá influenciar a decisão dos pais de optar por uma interrupção de gravidez.
No contexto da observação clínica, a apreciação das anomalias morfológicas faciais assume uma importância muito particular. Assim, em presença de uma criança dismórfica, o aspecto facial pode identificar uma determinada doença, reconhecer outra já vista anteriormente, mas não imediatamente identificável, ou simplesmente revelar uma situação completamente nova para nós. Nas situações difíceis, a comparação com outros casos publicados, o recurso a programas informatizados de diagnóstico diferencial com imagem, e a discussão clínica com outros colegas com experiência em dismorfologia, poderão ser de grande utilidade. Como noutras áreas da Medicina é preciso conhecer para diagnosticar.
Convém ter sempre presente que, se por um lado, um diagnóstico correcto tem todas as vantagens não só em termos de uma adequada intervenção terapêutica como na dispensa de exames desnecessários, por outro lado um diagnóstico errado, por falta de experiência ou precipitação, pode ter consequências muito graves. Rotular uma criança com um diagnóstico que não corresponde à sua situação invalida uma eventual intervenção terapêutica, multiplica múltiplas consultas e exames desnecessários e pode influenciar erradamente um casal quanto à sua vida reprodutiva. As consequências podem ser, pois, muito negativas.
Nunca é demais salientar um aspecto que nos parece muito importante e tem certamente forte repercussão no aconselhamento genético aos pais e na decisão quanto a futuras gravidezes. Trata-se do empenho que deve ser posto no esclarecimento etiológico de um feto ou de um recém-nascido com uma situação malformativa muito grave mesmo quando a morte pareça ser inevitável. O que parece ser inútil revela-se extremamente útil para o futuro.
O diagnóstico pré-natal, já abordado, noutro capítulo, tem tido nos últimos anos um grande desenvolvimento como método de prevenção secundária de anomalias congénitas. Mas, se por um lado as anomalias que estiveram na origem da interrupção médica de gravidez necessitam de ser comprovadas, por outro tem-se verificado um enorme interesse dos pais em saber as causas da morte fetal e o grau de risco para futuras gravidezes. Isto levou ao desenvolvimento de uma actividade multidisciplinar que é a embriofetopatologia clínica. Esta actividade, ponto de encontro de patologistas, dismorfologistas, geneticistas, perinatologistas e obstetras, no contexto dos Centros de Diagnóstico Pré-natal, tem protocolos próprios. Se em linhas gerais são semelhantes aos descritos no protocolo anterior, para a avaliação clínica dos nados-vivos, revestem-se, como é óbvio, de alguns aspectos particulares.
Assim, mantêm-se os 5 primeiros pontos, com excepção naturalmente do neurodesenvolvimento, bem como do ponto 7. No que respeita ao ponto 6, está provado que a tentativa de efectuar estudo citogenético após a morte tem taxas de sucesso baixas e muito dependentes das condições em que as colheitas são realizadas.
Daí que é da maior importância enquanto o feto está vivo, colher e armazenar produtos biológicos para estudos de biologia molecular, bioquímicos ou outros, que estão naturalmente comprometidos quando existe morte fetal, embora no caso da biologia molecular seja possível utilizar material fetal obtido em certas condições para armazenamento de ADN. Torna-se necessário, portanto, desenvolver protocolos de participação entre os especialistas acima referidos, de forma a tornar possível o diagnóstico da causa de morte fetal e o aconselhamento genético aos pais.
Registos Nacionais e Internacionais
Existem, actualmente, em muitos países registos da ocorrência e natureza das AC bem como das circunstâncias pessoais, familiares e ambientais do seu aparecimento. Estes registos têm como objectivo a determinação da prevalência nacional e regional das AC e a determinação das suas causas.
Em Portugal, além de alguns Registos regionais ou de Registos nacionais por patologias, habitualmente sediados em Serviços Hospitalares, existiu um Registo Nacional de AC da responsabilidade do Instituto Nacional de Saúde (Centro de Estudos e Registo de Anomalias Congénitas – Cerac), que teve o seu início em 1996.
Actualmente, também na dependência do INSA/Departamento de Epidemiologia, existe o chamado RENAC (Registo Nacional de Anomalias Congénitas) que, tal como o anterior CERAC, é um registo de base populacional que recebe notificações de várias origens, principalmente dos Serviços Hospitalares de Obstetrícia, Pediatria e especialidades pediátricas, mas também de outros Serviços como Anatomia Patológica e Genética Médica. Os seus objectivos consistem em determinar a prevalência das AC e a sua distribuição geográfica por residência das mães, observar as suas variações e tendências espaciais e temporais e estabelecer um sistema de vigilância epidemiológica.
São notificados todos os recém-nascidos vivos cujas anomalias sejam detectadas até ao final do período neonatal, as mortes fetais com anomalias e as interrupções de gravidez por patologia malformativa. São registadas as anomalias estruturais major, mas não as minor quando isoladas. (ver adiante)
Segundo o relatório do RENAC, abrangendo um período de 11 anos (2000-2010 a que correspondem 1.298.580 nados-vivos em Portugal), foram notificados 11.502 casos e diagnosticadas 17.502 AC. Em 72,6% dos RN observou-se uma AC isolada e, em 27,4%, AC múltiplas.
As AC cardiovasculares foram as mais prevalentes com 38,8 casos/10.000 nascimentos, seguindo-se-lhes as AC musculoesqueléticas (29,09 casos /10.000 nascimentos, e as do aparelho urinário (com 19,29/10.000 nascimentos). As AC de origem cromossómica surgiram com uma prevalência de 13,42/10.000 nascimentos.
Na Europa existem outros Registos de AC, nacionais ou regionais. O EUROCAT (European Registry of Congenital Anomalies and Twins) é um Projecto financiado pela Comissão Europeia, constituído por uma rede de vários Registos regionais europeus que trabalham com a mesma metodologia e publicam os seus dados em conjunto. Portugal colabora no Eurocat desde 1990 com a Região a sul do Tejo.
É ainda de assinalar a existência de um importante Registo com uma participação populacional muito mais alargada, a International Clearinghouse for Birth Defects Monitoring Systems, que reúne vários países da Europa, Ásia e Américas do Norte, Centro e Sul.
Prevenção
Num contexto global da prevenção cabe aos profissionais de saúde que trabalham na comunidade um papel muito importante. O seu conhecimento da patologia familiar, das condições ambientais porventura perigosas em que decorre a vida das famílias e o papel que desempenham nas consultas de planeamento familiar, tornam-nos interlocutores privilegiados no contexto das actividades que contribuem para a prevenção das anomalias congénitas.
Se, pelo conhecimento do contexto familiar, os mesmos podem identificar anomalias ou síndromas hereditárias e situações de risco durante a gravidez e providenciar o recurso a consultas especializadas, por outro lado podem ter um papel decisivo na prevenção primária de algumas situações frequentes, mas evitáveis.
Assim, as embriopatias ocasionadas pela diabetes materna e pela rubéola, a embriofetopatia alcoólica e os defeitos do tubo neural, são exemplos destas situações nas quais o controle adequado da diabetes materna, a vacinação anti-rubéola em tempo útil, o combate aos hábitos alcoólicos da mulher na idade reprodutiva e a administração de ácido fólico no período pré-concepcional, são medidas decisivas para diminuir a morbilidade e a mortalidade de algumas anomalias congénitas.
A prevenção de algumas anomalias congénitas é, pois, possível, mas seguramente exige um trabalho colectivo.
BIBLIOGRAFIA
Cassidy SB, Allanson JE. Management of Genetic Syndromes. Hoboken, NJ-USA: Wiley-Liss, 2005
Davies DP, Evans DJR. Clinical dysmorphology: understanding congenital abnormalities. Curr Pediatr 2003;13: 288-297
Encha-Razavi, Escudier E. Embryologie humanaine, de la molécule à la clinique. Paris: Masson, 2001
Epstein CJ, Erickson RP, Winshaw-Boris A (eds). Inborn Errors of Development: The Molecular Basis of Morphogenesis. New York: Oxford University Press, 2004
Eurocat. Report 8: Surveillance of Congenital Anomalies in Europe, 1980-1999. University of Ulster, 2002. www.eurocat.ulster.ac.uk (acesso em Março 2012)
Feijoó MJ. Dismorfologia Clínica. In Palminha JM e Carrilho EM (eds). Orientação Diagnóstica em Pediatria. Lisboa: Lidel, 2002
Ferkol TW, Leigh MW. Ciliopahies: the central role of cilia in a spectrum of pediatric disorders. J Pediatr 2012; 160: 366 – 371
International Clearinghouse for Birth Defects Monitoring Systems. World Atlas for Birth Defects. Geneva: WHO, 2003
Jones KL. Smith’s Recognizable Patterns of Human Malformation. Philadelphia: Saunders, 2013
Kliegman RM, Stanton BF, StGeme JW, Schor NF (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier Saunders, 2015
Laranjeira A, Clington A, Carvalhosa G, Henriques M, Amaral JMV. Anomalias congénitas em 30.625 nascimentos consecutivos. Arq do H D Estefânia 1990; 5:159-164
Martinez-Frias ML, Frias JL, Opitz JM. Errors of morphogenesis and development field theory. Am J Med Genet 1998; 76: 291-296
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Nunes L, Carvalho MCA. A contribuição das malformações congénitas para a mortalidade infantil em Portugal 1991-99. Saúde Infantil 1995;17: 47-52
OMIM-Online Mendelian Inheritance in Man. www.ncbi.nlm.nih.gov/entrez (acesso em Março, 2018)
Larsen WJ. Essentials of Human Embriology. New York: Churchil Livingstone, 2008
Opitz JM. The development field concept in clinical genetics. J Pediatr 1982; 101:805-809
Opitz JM, Zanni G, Reynolds Jr JF, Gilbert-Barness E. Defects of blastogenesis. Am J Med Genet 2002; 115: 269-286
Park SM, Marthur R, Smith GCS. Congenital Anomalies After Treatment for Infertility. BMJ 2006; 333: 665-666
Registo Nacional de Anomalias Congénitas/ RENAC. Relatório de 2000-2010. Lisboa: Instituto Nacional de Saúde, 2010
Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA (eds). Rudolph´s Pediatrics. New York: McGraw-Hill Medical, 2011
Spranger J, Bernirschke K, Hall JG, et al. Errors of morphogenesis: Concepts and terms. J Pediatr 1982; 100: 160-165
www.orpha.net
www.rarediseases.org
www.emedicine.com/ped
www.genetests.org
www.marchofdimes.com/peristats
(acesso em Junho, 2018)
AGRADECIMENTOS
Agradecemos à Unidade de Fetopatologia do Hospital de Egas Moniz toda a colaboração iconográfica do presente capítulo.