*Revisão de Aguinaldo Cabral
Sistematização
As doenças hereditárias do metabolismo (DHM) dos aminoácidos compreendem diferentes situações clínicas como: hiperfenilalaninémias (incluindo fenilcetonúria); tirosinémia dos tipos I, II e III; alcaptonúria, hawkinsinúria; acidémias/acidúrias orgânicas de cadeia ramificada (leucinoses, acidémias propiónica, metilmalónica, isovalérica e outras); doenças do ciclo da ureia; doenças dos aminoácidos sulfurados (incluindo homocistinúria e outras); hiperornitémias, síndroma HHH; acidúrias orgânicas “cerebrais” e doenças do catabolismo da lisina (incluindo a acidúria glutárica tipo I, a L2-hidroxiglutárica, a D2 – hidroxiglutárica, a acidúria N-acetilaspártica ou doença de Canavan); hiperglicinémia não cetótica; doenças do metabolismo da prolina e da serina; defeitos do transporte dos aminoácidos através das membranas celulares (cistinúria, intolerância proteica lisinúrica, doença de Hartnup) e muitas outras.
Etiopatogénese
As deficiências de determinadas enzimas envolvidas no metabolismo dos aminoácidos conduzem frequentemente a sinais e sintomas de intoxicação aguda ou crónica por acumulação de metabólitos e lesão tecidual. Os órgãos mais frequentemente afectados são o sistema nervoso central, o fígado e os rins.
A expressão clínica e da gravidade dependem fundamentalmente do grau de deficiência enzimática e, particularmente, da ingestão proteica e da produção endógena decorrente do catabolismo proteico.
Manifestações clínicas
A idade de apresentação é variável.
No período neonatal, geralmente após um intervalo livre que pode ser inferior a 24 horas, depois do início da alimentação, ocorrem os seguintes sinais, por vezes associados: recusa alimentar, sucção pobre, episódios de apneia, vómitos, não ganho de peso, hipotonia, letargia, convulsões, hipotermia, coma, alterações do tono muscular, mioclonias e, por vezes, odor anómalo.
Após o período neonatal, as formas de apresentação podem ser: crises agudas ou recorrentes de coma, vómitos crónicos, acidose, hipoglicémia, ataxia, alterações do comportamento, neuropatia, deterioração neurológica e/ou mental progressiva, autismo, etc..
Na primeira infância, coincidindo com a diversificação alimentar, poderão surgir febre, anorexia ou vómitos.
Na puberdade, o crescimento e eventuais alterações do foro endócrino poderão constituir factores desencadeantes de estresse metabólico.
Exames complementares
Perante a suspeita de doença relacionável com defeitos do metabolismo dos aminoácidos, e paralelamente às análises em amostras de sangue, urina e LCR, discriminadas no capítulo anterior, devem ser feitas outras análises mais específicas em laboratório especializado, a partir das referidas amostras:
- Cromatografia dos aminoácidos no sangue e urina;
- Cromatografia dos ácidos orgânicos na urina;
- Cromatografia dos aminoácidos no LCR se suspeita de encefalopatia metabólica;
- Perfil de acilcarnitinas no plasma.
Outros exames a solicitar dependerão da suspeita diagnóstica específica (ver adiante).
Tratamento de emergência
Em muitas destas situações verifica-se descompensação aguda directamente relacionada com incremento da ingestão proteica ou com estado catabólico, factor altamente deletério.
Assim, as principais linhas de actuação incluem:
- Interromper o estado catabólico: propiciando um suprimento energético aumentado, geralmente soluto glicosado endovenoso (excepcionalmente por sonda nasogástrica), se necessário associando a administração de insulina por via endovenosa.
- Interromper o suprimento proteico: propiciando a chamada “nutrição de emergência” através de nutrição (entérica ou parentérica de acordo com a situação clínica) à custa de hidratos de carbono, lípidos, NaCl, KCl, gluconato de cálcio e água durante 24-48 horas.
Após este período, o suprimento proteico é iniciado cautelosamente de modo progressivo com leite materno, se possível, ou com fórmula; se a via entérica não for viável, procede-se à nutrição parentérica com solução de aminoácidos em incrementos progressivos; nesta última circunstância, com a melhoria do quadro clínico, procede-se à transição para a alimentação entérica, geralmente dentro do período de 4-5 dias. - Propiciar suprimento hídrico e electrolítico adequados. Níveis séricos de sódio dentro dos limites da normalidade reduzirão o risco de edema e lesão cerebrais.
- Terapêutica medicamentosa específica ou vitamínica (cofactores enzimáticos) dependendo da patologia em causa (por exemplo, biotina na acidúria propiónica, hidroxicobalamina na acidúria metilmalónica, carnitina nas acidúrias orgânicas, glicina na acidúria isovalérica, etc.), e fármacos que promovem a eliminação da amónia (benzoato de sódio, fenilbutirato de sódio nas doenças do ciclo da ureia).
- Meios de depuração dependendo da patologia, estado clínico e achados laboratoriais: diurese forçada, diálise peritoneal, hemodiálise, hemofiltração, exsanguinotransfusão, etc..
Tratamento de manutenção
- Dietético: dieta hipoproteica, e suplemento de mistura específica aminoácidos (não contendo os aminoácidos cuja via metabólica está bloqueada) associado a oligoelementos e sais minerais.
De referir que a utilização de mistura de aminoácidos é crucial nos casos de fenilcetonúria, leucinose, tirosinémia, homocistinúria, e no defeito da ornitina-aminotransferase; noutras situações, como as acidúrias orgânicas, a mistura de aminoácidos é mais controversa, especialmente na fase aguda, podendo contribuir para elevação da amónia. No entanto, cabe referir que a restrição proteica excessiva pode levar a catabolismo. - Medicamentoso: aplicam-se as noções referidas a propósito do tratamento de emergência que contribuam para a desintoxicação, por exemplo, nas doenças do ciclo da ureia, tirosinémia tipo I, etc..
Avaliação regular do crescimento, desenvolvimento e parâmetros laboratoriais
Chama-se a atenção para o risco de má-nutrição e de deficiências nutricionais diversas como resultado do regime dietético restritivo.
De acordo com as patologias em causa, para além das análises para avaliação global, haverá que incluir o ionograma sérico, ureia no sangue e urina, determinação de proteínas totais e fracções, amoniémia, aminoácidos urinários e plasmáticos e ácidos orgânicos na urina. Importa igualmente a avaliação nutricional.
Educação da família
A família deve ser instruída sobre as características principais da doença em causa, sobre a sintomatologia nas crises de descompensação, sobre critérios de gravidade e sobre medidas emergentes a tomar. Torna-se, assim, fundamental que a família aprenda a contactar de imediato o centro especializado de tratamento a que a criança deve recorrer nas situações graves.
No âmbito deste capítulo é feita uma referência especial à fenilcetonúria, à tirosinémia do tipo I, à homocistinúria, à leucinose, às acidúrias orgânicas clássicas e às doenças do ciclo da ureia.
1. FENILCETONÚRIA
Esta doença, vulgarmente designada nos países de língua inglesa pela sigla PKU (de phenylketonuria), pertence ao grupo das hiperfenilalaninémias.
A PKU na sua forma clássica surge com uma frequência estimada entre 1/10.000 e 1/20.000 RN; em Portugal, até final do ano de 2005, em 2.590.890 RN foi encontrada prevalência ~1/11.000 e, em 2015, ~1/12.150.
A mesma resulta do défice total ou parcial da enzima fenilalanina – hidroxilase hepática (PAH) originando níveis elevados de fenilalanina e seus metabólitos no sangue; o gene que codifica a fenilalanina-hidroxilase localiza-se no cromossoma 12q24.1, descrevendo-se uma diversidade de mutações (mais de 500). A maioria dos doentes corresponde a heterozigotias para dois diferentes alelos mutantes.
De referir que em cerca de 1-3% dos casos com valores elevados de fenilalanina no sangue, a anomalia é explicada por défice duma das enzimas necessárias para a produção ou renovação do cofactor tetra-hidro-biopterina (BH4); trata-se das chamadas formas malignas de hiperfenilalaninémia, ou mais correctamente, hiperfenilalaninémias por defeito de BH4, muito graves, não respondendo à dieta hipoproteica isolada.
Quanto a manifestações clínicas, salienta-se que a criança afectada é assintomática na data do nascimento mas, caso não se verifique qualquer intervenção (o que acontecia na era pré-rastreio) verifica-se atraso do neurodesenvolvimento progressivo e grave: atraso psicomotor, da locomoção, da fala, hiperactividade frequente, comportamento autista, negativismo, etc.. Pode igualmente verificar-se quadro de hipsarritmia (“espasmos infantis”), hipertonia e hiperreflexia osteotendinosa; sem tratamento, surge deterioração neurológica e mental progressiva.
Os fenilcetonúricos podem apresentar, de modo inconstante, um fenótipo clínico particular: pele clara, dermatose aparentando eczema, cabelos loiros e olhos azuis; trata-se dum pseudo-albinismo secundário.
O tratamento dietético é fundamental: consiste numa dieta hipoproteica, semi-sintética, suplementada com aminoácidos apropriados, sendo o suprimento em fenilalanina reduzido e controlado.Tal regime deve ser mantido durante toda a vida.
O leite materno pode ser usado durante os primeiros meses de vida, sob rigorosa vigilância do centro especializado de tratamento.
A vigilância metabólica é feita com o doseamento regular do nível sérico da fenilalanina, o qual deve ser mantido entre 3-6 mg/dL. Chama-se, a propósito, a atenção para o facto de dietas extremamente restritivas, originando valores séricos < 3 mg/dL, poderem conduzir a quadros de défice de fenilalanina (que é um aminoácido essencial) traduzido por hipocrescimento, letargia, anemia, alterações ósseas e até, morte.
Estão descritas formas de hiperfenilalaninémia moderada, benigna, com valores de fenilalaninémia ligeiramente elevados, até 6-6,5 mg/dL, com dieta normal. Nestes casos o prognóstico parece ser bom, sem necessidade de regime alimentar restritivo, sendo, no entanto, prudente o seguimento clínico (com atenção especial ao exame neurológico) e a vigilância laboratorial periódica.
Alguns centros utilizam um biomarcador independente dos níveis sanguíneos fenilalanina: a ADMA (dimetilarginina assimétrica). Na prática, utiliza-se a ratio ADMA/creatinina, com interesse sobretudo para detectar casos de pacientes com dieta eventualmente não balanceada, susceptível de efeitos negativos a longo prazo.
Se o indivíduo afectado atingir a idade adulta e, no sexo feminino, a idade de procriar, há que atender a que níveis elevados de fenilalanina durante a gravidez podem ter efeito lesivo sobre o feto (aborto frequente, baixo peso de nascimento, restrição do crescimento fetal, defeitos cardíacos, microcefalia e outras anomalias congénitas). É necessário, pois, que a mulher com hiperfenilalaninémia cumpra dieta muito rigorosa antes da concepção e durante toda a gravidez, de modo a manter níveis séricos de fenilalanina entre 1-3 mg/dL.
As novas terapias incluem:
- Suplementação de aminoácidos neutros, os quais competem com o transporte da fenilalanina/Phe;
- Administração de BH4 (sapropterina di-hidrocloreto- Kuvan) em doentes seleccionados. A resposta/sensibilidade ao BH4 necessita da presença de alguma actividade residual da PAH. Permite aumentar, duas a três vezes, a tolerância alimentar à PHE, com menores ou nulas restrições dietéticas;
- Terapia enzimática com PAL (fenilalanina – amónia – liase), proteína que converte a Phe em excesso em metabólito não tóxico;
- Administração de glicomacropeptídeo, proteína natural derivada da caseína resultante do fabrico de queijo. Está presente no soro do leite e, na forma pura, não contém Phe. Os alimentos preparados a partir desta proteína são boa alternativa à mistura de aminoácidos sintéticos;
- Alimentos hipoproteicos de nova geração e novas apresentações de misturas de aminoácidos em barras, saquetas, comprimidos, líquidos são resultado dos enormes progressos da tecnologia;
- – Terapia génica, que se tem desenvolvido na procura de correcção duradoira ou permanente do fenótipo PKU.
2. TIROSINÉMIA DO TIPO I
A tirosinémia do tipo I (ou tirosinémia hepatorrenal) é uma doença autossómica recessiva rara, provocada por défice da enzima fumaril-aceto-acetato-hidrolase originando elevação da tirosina sérica e acumulação de metabólitos tóxicos intermediários.
Como consequência surge compromisso grave ao nível do fígado, rim e nervo.
A doença raramente tem início no período neonatal; dum modo geral, surge a partir da 4ª-5ª semana de vida e, mais frequentemente, nos primeiros meses.
Trata-se duma doença hepática grave, com insuficiência hepática aguda que pode ser fatal. Os sinais clínicos poderão ser desencadeados por doença intercorrente levando a estado catabólico (por ex. febre).
Caracteriza-se por icterícia, hepatosplenomegália, edema, diátese hemorrágica, ascite, hipoglicémia, e, por vezes, odor a “couve cozida”. Pode existir disfunção tubular renal complexa, raquitismo de causa renal, e, raramente, sinais de polineuropatia periférica aguda (simile porfíria). Os doentes estão em risco de sofrer de hepatocarcinoma, o qual pode aparecer precocemente. (Figura 1)
No âmbito da avaliação laboratorial do quadro sindrómico de hepatopatia e diátese hemorrágica, cabe referir níveis elevados de ALT e AST traduzindo citólise, e níveis baixos dos factores de coagulação II, VII, IX, XI, XII.
O diagnóstico diferencial faz-se fundamentalmente com a galactosémia e intolerância hereditária à frutose pela hepatopatia, diátese hemorrágica e tubulopatia.
O diagnóstico baseia-se na demonstração de níveis elevados de tirosina e de alfa-fetoproteína (no sangue), de ácido aminolevulínico (ALA) (na urina), e da presença de succinilacetona (SA) na urina e sangue.
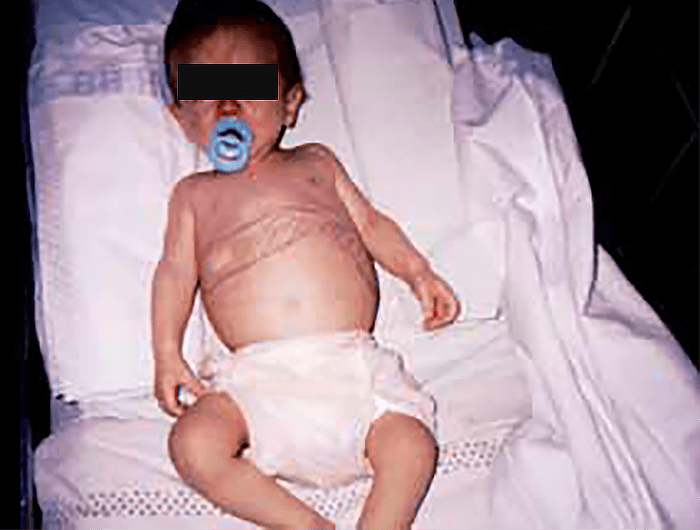
FIGURA 1. Lactente com quadro clínico de tirosinémia tipo I: hepatosplenomegália, sinais de raquitismo, e desnutrição com hepatocarcinoma aos 11 meses de idade. (Cortesia do Dr. Aguinaldo Cabral)
Salienta-se que esta última constitui melhor marcador para o diagnóstico do que a hipertirosinémia, a qual pode acompanhar outros tipos de hepatopatias agudas adquiridas.
Progressos recentes relacionados com o PNDP alargado permitem diagnóstico mais rápido com a medição da SA no sangue fresco.
A confirmação diagnóstica faz-se pela determinação da actividade da enzima acima referida em amostras de biópsia hepática, de culturas de fibroblastos, e ainda através de estudo genético (análise mutacional).
O tratamento consiste essencialmente:
- numa dieta hipoproteica com suprimento reduzido e controlado de fenilalanina e tirosina, suplementado com mistura apropriada de aminoácidos; e
- na utilização imediata do fármaco NTBC (nitro-triflurometil-benzoil-cicloexanediona), tricetona que inibe a hidroxifenil-piruvato dioxigenase, bloqueando a montante o catabolismo da tirosina, e evitando a acumulação de metabólitos tóxicos para o fígado, rim e nervo, o que se traduz numa melhoria dramática.
Trata-se duma terapêutica de primeira linha em qualquer idade, inclusivamente no RN em coma. Se não houver resposta ao NTBC (o que pode ocorrer em cerca de 10% dos casos) e/ou houver suspeita de malignidade hepática, o transplante do fígado impõe-se com urgência.
3. HOMOCISTINÚRIA
A homocistinúria clássica ou de tipo I, devida a deficiência da cistationina-beta-sintetase (CBS), é o erro inato do metabolismo da metionina mais frequente. Tal deficiência, relacionada com gene localizado no cromossoma 21q.22.3, leva à acumulação nos tecidos de metionina, homocistina e derivados, com perda de cistationina e baixa concentração de cistina.
No que respeita a manifestações clínicas, cabe referir que a doença está associada a anomalias graves em quatro órgãos ou sistemas: o olho (luxação do cristalino, miopia, glaucoma, etc.), o esqueleto (dolicostenomélia, aracnodactilia, escoliose, osteoporose, fracturas patológicas, mobilidade articular diminuída, etc.), o sistema nervoso central (insuficiência mental, AVC, sintomas psiquiátricos, etc.) e o sistema vascular (tromboembolismo das artérias e veias, a principal causa de morbilidade e mortalidade).
A acumulação de homocistina é provavelmente determinante do dano vascular generalizado e complicações tromboembólicas.
A criança é assintomática ao nascer, mas, se não for tratada, surgirá progressivamente o quadro clínico completo, incluindo o fenótipo semelhante ao da síndroma de Marfan (estatura elevada, ossos longos finos e alongados) e aracnodactilia na transição para a puberdade. Contudo, a restrição da mobilidade articular contrasta com a laxidão da autêntica síndroma de Marfan. As anomalias surgem significativamente mais cedo nos doentes não respondentes à piridoxina (vitamina B6). Casos moderados poderão ser reconhecidos após o surgimento de complicações tardias como os AVC.
O diagnóstico inicial assenta no perfil típico da cromatografia dos aminoácidos no plasma: elevação da metionina e homocistina, baixo nível de cistina, e não aumento de cistationina. A determinação da homocisteína total (tHcy) no plasma (p) é de grande valor diagnóstico, considerando-se valor normal de tHcy p: < 15 µmol/L; os doentes não tratados têm valores > 200 µmol/L.
O diagnóstico definitivo far-se-á determinando a actividade enzimática nos fibroblastos e hepatócitos (biópsia hepática) e por análise mutacional.
O tratamento tem como objectivo reduzir os níveis elevados de homocistina para valores próximos do normal, mantendo um ritmo de crescimento normal. O mesmo consiste em dieta com suprimento de metionina reduzido, e de cistina elevado, associando piridoxina, ácido fólico, vitamina C, vitamina B12 e betaína, em várias combinações; por vezes é necessária uma mistura específica de aminoácidos isenta de metionina.
Cerca de 50% dos doentes respondem, por vezes parcialmente, a doses altas de vitamina B6 (mas sempre < 1 grama/dia). Salienta-se igualmente a importância do tratamento antitrombótico. A ausência de resposta à piridoxina poderá estar relacionada com a carência em folato.
4. LEUCINOSES
As leucinoses são doenças do catabolismo dos aminoácidos de cadeia ramificada (AACR): leucina, isoleucina e valina, por deficiência do complexo da desidrogenase dos α-cetoácidos de CR (BCKD), complexo composto por três unidades catalíticas e duas enzimas reguladoras, codificadas por 6 loci genéticos diferentes: E1 (E1α e E1β), E2, E3, e as reguladoras BCKD-fosfatase e cinase.
Sobre a etiopatogénese, importa referir que a deficiência da BCKD origina uma elevação marcada dos AACR e dos seus cetoácidos no plasma, urina e LCR. Da isoleucina elevada forma-se, por racemização não enzimática, a alo-isoleucina, cuja presença é sistemática nos doentes com leucinose. Os AACR representam aproximadamente 40% das necessidades em AAE (essenciais).
O excesso de AACR e dos seus cetoácidos interfere com o metabolismo do sistema neurónio/astrócito, o que afecta a biossíntese das aminas biogéneas e altera o equilíbrio dos ciclos leucina/glutamato e glutamato/glutamina, no cérebro. A leucina e o ácido 2-cetoisocapróico elevados provocam disfunção cerebral: são os metabólitos mais neurotóxicos.
No que respeita à genética e epidemiologia, importa referir que se trata duma doença AR, pan-étnica, com incidência de 1/120.000 a 1/500.000 na Europa, sendo no Mundo à volta de 1/185.000 nados-vivos. Em Portugal é frequente nas comunidades ciganas, especialmente no sul do país.
Existem 6 formas de apresentação clínica: 1- Forma clássica: grave, de início neonatal; 2- Forma intermédia; 3- Forma intermitente; 4- Forma sensível à tiamina; 5- Forma por deficiência de E3; e 6- Formas assintomáticas, estas últimas na actualidade mais frequentemente diagnosticadas através da realização do rastreio alargado.
A Forma clássica, representando 80% das leucinoses, é caracterizada por encefalopatia + cetose. No geral, trata-se de um RN de termo que nasce bem, mas que, após um intervalo livre, seja entre o 4º e o 7º dia, evidencia sucessivamente: sucção débil, recusa alimentar, letargia, soluços, hipotonia, bradicardia, crises de apneia, hipotonia axial com hipertonia dos membros, movimentos de boxage e pedalagem, por vezes elevação lenta dos membros (espontânea ou após estimulação), tremores, opistótono, mioclonias, fontanela anterior hipertensa, um odor especial da urina (açúcar/ caramelo). Os abalos mioclónicos podem ser interpretados como convulsões; o EEG pode registar um padrão periódico (do tipo burst suppression).
A alimentação com leite materno pode atrasar os sintomas para a segunda semana de vida.
O odor característico (com algum valor na suspeição) pode surgir em RN alimentado com leite materno no contexto de mãe lactante tendo ingerido caril, especiarias, condimentos, ou de o coto umbilical ter sido limpo com iodopovidona.
Sem tratamento, rapidamente surge o coma e morte precoce. Durante o coma podem ser observados sinais neurológicos focais, hemiplegia aguda, hemianópsia e sinais de edema cerebral.
Para o diagnóstico, torna-se fundamental a realização dum conjunto de exames, predominantemente bioquímicos e, complementarmente, imagiológicos.
A cromatografia de aminoácidos (CAA) no plasma, sangue, urina e LCR mostra valores elevados de AACR (especialmente de leucina), dos respectivos cetoácidos (em especial do 2-cetoisocapróico) e de alo-isoleucina.
O teste DNPH (di-nitro-fenil-hidrazina) na urina, método colorimétrico detectando os cetácidos de CR, é positivo na fase aguda.
A presença de alo-isoleucina no plasma e urina é patognomónica, mas poderá não surgir até ao 6º dia de vida.
Existe cetonúria acentuada e a amónia pode estar elevada, embora com valor < 130 µmol/L). Mais raramente pode ocorrer acidose e hipoglicémia. A razão alo-isoleucina/ isoleucina > 0,6 é típica da Forma Clássica.
Com valores de leucina plasmática > 800 µmol/L, a encefalopatia torna-se evidente.
Através da imagiologia cerebral (TAC, RM) podem ser observados sinais de edema cerebral generalizado e de alterações da substância branca profunda: cerebelo, pedúnculos e cápsula interna. Salienta-se, pois, que a leucinose é uma doença da substância branca.
Para a confirmação diagnóstica procede-se à determinação da actividade enzimática nos fibroblastos/ linfoblastos em cultura, ou a estudos de biologia molecular.
O diagnóstico pré-natal é possível.
As complicações são frequentes: edema cerebral, sobretudo no RN e lactente; nos doentes mais velhos poderá surgir compressão do tronco cerebral e morte súbita (atenção à reidratação vigorosa). Outras complicações: desmielinização, alopécia, descamação cutânea, ulcerações da córnea, pancreatite e desnutrição; esta última poderá ser reversível com dieta correcta e continuada.
I – O tratamento do RN sintomático com a forma clássica é uma emergência médica.
Ia – Na fase aguda
Os objectivos são: reduzir rapidamente os níveis plasmáticos de AACR; combater o catabolismo; fomentar o anabolismo; providenciar o suprimento adequado de nutrientes. Assim:
- fluidoterapia IV (soro fisiológico se necessário), mantendo valores plasmáticos de Na > 140 mEq/L e Osmol > 290 mOsm/L, com atenção ao edema cerebral, e evitando glicémia > 150-200 mg/dL;
- métodos dialíticos (HFvv, HD – sendo que os AACR têm baixa depuração/ clearance renal), de acordo com os seguintes critérios: sintomas neurológicos graves, leucina (p) > 1500 µmol/L, intolerância gástrica (frequente), descida de leucina (s) < 500 µmol/L após 24 horas de dieta específica, ausência de melhoria clínica;
- paragem de proteína natural (24-48 horas); suprimento energético > 100 kcal/kg/dia;
- introdução de mistura de aminoácidos (Mx AA) livre de AACR para aumentar a síntese proteica;
- suplementos de valina e isoleucina, ao 2º-3º dia de terapia, na dose 300-400 mg/dia de cada, para fomentar a síntese proteica e evitar a sua deficiência precoce, visto se eliminarem mais rapidamente do que a leucina; a dieta será administrada, consoante os casos, por sonda nasogástrica ou por alimentação parentérica total;
- tiamina: 100-500 mg/dia, particularmente nas formas sensíveis à vitamina B1; alguns centros advogam uma terapia dietética apenas (evitando a diálise), mas somente se o diagnóstico for precoce (antes do 3º-7º dia de vida) e em doentes assintomáticos.
- após as 24-48 horas sem proteínas naturais, deve iniciar-se um suprimento cauteloso das mesmas, incluindo de leucina, até à tolerância do doente.
Ib – Na fase de manutenção
Os objectivos são: manter o equilíbrio metabólico e alcançar um bom estado nutricional, de crescimento e de desenvolvimento. Assim:
- dieta hipoproteica, semissintética, regularmente ajustada, para toda a vida. Os suprimentos de leucina são monitorizados de acordo com os respectivos níveis plasmáticos;
- manter a ingestão de Mx de AA livre de AACR, que é crucial (pode fornecer-se na proporção de ~90% da proteína total);
- manter o suplemento de valina e isoleucina até ser necessário;
- manter o suplemento de tiamina (é prática comum);
- suprimentos de leucina cerca de 300-400 mg/dia (60 a 110 mg/kg/dia) até aos 6 meses de idade. Crianças maiores de 3 anos, adolescentes e adultos podem tolerar até 500-700 mg/dia;
- vigiar/corrigir possíveis deficiências de: isoleucina, valina, cálcio, magnésio, zinco, selénio e outros, e também de ácidos gordos (AG) essenciais;
- usar tabelas de equivalentes: 1 parte é igual ao peso em gramas de um alimento que forneça 50 mg de leucina. Muito útil na confecção diária das dietas: evita monotonia alimentar e melhora o estado nutricional.
Nota importante:
|
Ic – Durante as intercorrências
Se no decurso da terapia ocorrerem situações febris, vómitos, diarreia (que aumentam o catabolismo), os níveis plasmáticos dos AACR e cetoácidos podem atingir níveis neurotóxicos em poucas horas, surgindo então sinais de alarme: apatia, ataxia, alucinações, anorexia, convulsões, alterações do equilíbrio e do comportamento.
Os pais devem procceder de imediato à realização do teste DNPH na urina para detectar a cetonúria, reduzir/ suprimir o suprimento de proteína natural, fornecer mais energia, manter os suplementos de Mx de AA, de valina e de isoleucina. Se houver intolerância gástrica, e ao mínimo sinal de alteração da consciência, deve contactar-se prontamente o centro de referência hospitalar.
II– Outras terapias
- transplante hepático: já executado com êxito: o risco de descompensação em eventos catabólicos parece abolido;
- transplante de hepatócitos: o interesse nos humanos está em avaliação;
- fenilbutirato: reduz a concentração plasmática dos AACR e cetoácidos, parecendo aumentar a activiade residual da BCKD;
- recentemente: introdução de fórmulas enriquecidas em vários AA que competem no transporte dos AACR através da BHE, diminuindo a entrada da leucina no cérebro;
- uso de norleucina: potente competidor da leucina ao nível da BHE, diminuindo a sua entrada e acumulação, e especialmente do 2-cetoisocapróico, no cérebro. É um isómero da leucina e isoleucina.
Nas variantes moderadas, se assintomáticas, não é clara a necessidade de terapêutica de longo prazo; contudo, os doentes e famílias devem saber evitar/ corrigir hipotéticas crises.
Nos casos de leucinose materna: dieta hipoproteica rigorosa; deve manter-se concentração de leucina entre 100-300 µmol/L, e de valina e isoleucina, normal a ligeiramente elevada. É possível o nascimento de filho saudável. Todavia, há que ter em atenção o puerpério: risco de descompensação metabólica da mãe até 6 a 8 semanas após o parto.
Sobre a evolução e prognóstico, há a salientar os seguintes factos:
- nos sobreviventes poderão surgir sequelas neurológicas graves, insuficiência cognitiva, espasticidade e cegueira cortical;
- actualmente verificam-se menor morbilidade, menor mortalidade e mais baixa proporção de hospitalizações. Mais de 1/3 dos doentes com a forma clássica alcançarão QI > 90, e 1/3 entre 70-90. Os doentes que apresentam espasticidade, quadriplegia, regra geral, têm pior prognóstico intelectual.
5. HIPERGLICINÉMIA NÃO CETÓTICA
A glicina é o aminoácido mais pequeno que existe, não essencial, actuando como neurotransmissor inibidor no SNC.
A doença mais representativa em relação com este aminoácido é a hiperglicinémia não cetótica a qual se deve a um defeito no complexo multienzimático encarregado de metabolizar a glicina e diminuir a sua concentração no organismo.
Existem duas formas de apresentação clínica:
- precoce, mais grave e frequente; e
- tardia.
A forma precoce (neonatal) cursa com encefalopatia grave e deterioração neurológica progressiva (hipotonia generalizada, convulsões e ulterior espasticidade); o prognóstico é muito reservado.
A forma tardia, podendo surgir em qualquer fase da infância ou no adulto, traduz-se essencialmente por movimentos paroxísticos coreicos, confusão mental e alterações do comportamento.
A glicina está elevada no plasma (p) e no LCR, com razão LCR/p elevada
(normal = < 0,02; na situação em análise: proporção muito elevada = > 0,08).
O tratamento com dextrometorfan, benzoato de sódio e folatos é de sucesso limitado.
6. ACIDÚRIAS ORGÂNICAS
SISTEMATIZAÇÃO
As acidúrias orgânicas (AO) são devidas a deficiências enzimáticas no metabolismo mitocondrial dos ácidos carboxílicos activados pela CoA, muitos dos quais resultam do catabolismo dos aminoácidos (AA).
São devidas, não só à acumulação de intermediários tóxicos, mas também à alteração do metabolismo energético mitocondrial e da homeostase da carnitina.
Incluem-se nas AO a deficiência de biotinidase ou de holocarboxilase sintetase levando a deficiência múltipla de carboxilases.
O termo mais correcto é acidúria, e não acidémia, porquanto se trata de doenças essencialmente detectadas pela análise da urina.
As AO mais conhecidas, ditas clássicas, incluem as seguintes nosologias:
Grupo I
- Acidúria propriónica (AP);
- Acidúria metilmalónica (AMM); e
- Acidúria isovalérica (AIV).
Grupo II
- Acidúria metilmalónica (AMM) e homocistinúria (forma especial de AMM).
Como exemplos de AO mais raras, não abordadas neste capítulo, citam-se: 3-metil-crotonil-glicinúria; |
Grupo I
As AP, AMM e AIV, ditas clássicas, são causadas por defeitos do catabolismo dos AACR (leucina, isoleucina, valina).
Formas de apresentação
- Forma de início neonatal, grave;
- Forma de início tardio, aguda, intermitente;
- Forma crónica, progressiva;
- Formas assintomáticas.
A forma de início neonatal comporta-se como encefalopatia metabólica de “tipo intoxicação” que surge após um intervalo livre de sintomas.
O início é marcado por deterioração (sem causa aparente e sem resposta à terapia sintomática). Primeiros sinais: recusa alimentar e sonolência, a que se seguem coma, desregulação neurovegetativa (dificuldade respiratória, soluços, episódios de apneia, bradicardia, hipotermia).
No coma é frequente ocorrerem alterações do tono muscular e movimentos involuntários, episódios de hipertonia generalizada com opistótono, movimentos de pedalagem e boxage, e outras manifestações de intoxicação central como hipotonia axial com hipertonia dos membros, tremores e espasmos mioclónicos. No EEG é comum um padrão periódico (burst-suppression). A desidratação é frequente, assim como hepatomegália moderada. Na AIV é dado importante o odor a pés suados.
As características bioquímicas incluem: acidose metabólica, cetose, hiato aniónico com valor elevado, hiperamoniémia de grau variável (se > 500 µmol/L poderá induzir alcalose respiratória e a suspeita errada de doença do ciclo da ureia), hipocalcémia, glicémia normal, baixa ou elevada (se elevada poderá induzir suspeita errada de coma diabético), neutropénia, anemia, trombocitopénia, pancitopénia (podendo induzir suspeita errada de sépsis) e, por vezes, valor do lactato elevado.
A forma de início tardio, de modo agudo e intermitente, surge geralmente após intervalo livre longo (por vezes superior a um ano), ou mesmo até na adolescência e na idade adulta.
Manifestações clínicas
As manifestações clínicas traduzem-se por crises recorrentes, intercaladas por períodos assintomáticos, como regra.
A crise inicial pode ser fatal; as recorrentes ocorrem, em geral, no contexto de infecção, estado catabólico, suprimento alimentar rico em proteínas, ou sem causa aparente. São comuns situações de coma recorrente (de todos os tipos) e crises recorrentes de ataxia e letargia.
Os doentes poderão apresentar sinais neurológicos focais, sintomatologia de edema cerebral (sugerindo erradamente encefalite, AVC ou tumor cerebral).
Outra apresentação traduz-se por um quadro similar ao da síndroma de Reye.
De salientar que a neutropénia e trombocitopénia podem ser sinais inaugurais.
A forma crónica, progressiva pode manifestar-se por anorexia persistente, vómitos crónicos, má progressão ponderal e osteoporose. Tal sintomatologia poderá levar à suspeita errada de: refluxo gastresofágico, intolerância às proteínas do leite de vaca, doença celíaca, ou de estenose pilórica tardia.
Noutros casos, verifica-se hipotonia, fraqueza muscular e massas musculares pobres sugerindo miopatia ou doenças neurológicas congénitas; noutros ainda, atraso de desenvolvimento não específico, atraso psicomotor progressivo, insuficiência mental, convulsões e patologia dos movimentos.
Frequentemente, os doentes permanecem por longo tempo sem diagnóstico correcto até à ocorrência de crise neurológica aguda e coma; nestas circunstâncias poderá então surgir a suspeita de DHM. |
As formas assintomáticas, descritas cada vez com maior frequência como resultado do rastreio alargado, podem ser representadas pela AIV associada a mutação C932T (A282V), geralmente em heterozigotia, e com fenótipo bioquímico moderado. Tais formas assintomáticas de AO levantam dúvidas quanto à necessidade de tratamento dietético contínuo e ao prognóstico a longo prazo.
Complicações
As complicações das AO clássicas são várias: sindroma extrapiramidal aguda ou progressiva, envolvimento ou mesmo necrose dos gânglios basais, atrofia cerebral, atraso de mielinização e alterações da motilidade.
Na AMM, de modo especial: falência renal que pode ocorrer pelos 10 anos de idade e progredir até à insuficiência renal terminal, necessitando de diálise e/ou TR; as lesões cutâneas são também comuns: descamação, alopécia, úlceras da córnea, geralmente devidas a má-nutrição proteica e deficiência de isoleucina.
Na AIV salienta-se: pancreatite aguda ou crónica que pode ser o quadro inaugural nos casos tardios, e ainda, cardiomiopatia.
Na AP pode surgir igualmente cardiomiopatia, a qual poderá estabelecer a indicação para transplante.
Diagnóstico
Quanto ao diagnóstico, importa referir que se torna fundamental a realização, entre outros, dos seguintes exames laboratoriais:
- cromatografia de AA plasmáticos (CAA);
- cromatografia de ácidos orgânicos urinários (CAO) podendo evidenciar o perfil específico de cada patologia;
- doseamento no plasma da carnitina total, livre e acilcarnitinas, amónia e lactato.
A acilcarnitina anómala na AP e AMM é a propionilcarnitina (C3), e na AIV a isovalerilcarnitina (C5).
A marca bioquímica típica das AO Clássicas é: acidocetose e hiperamoniémia secundária.
O diagnóstico deve ser confirmado por estudos enzimáticos ou por estudo genético mutacional.
Tratamento
O tratamento das AO clássicas (AP, AMM e AIV) inclui procedimentos diversos na fase aguda e na fase de manutenção
Na fase aguda
- combater o catabolismo com altas doses endovenosas de glucose (mais insulina, se necessário); corrigir a acidose e tratar a infecção e a anemia;
- interromper o suprimento de proteínas (máximo: 24-48 horas) a par de suprimento energético normal/ elevado;
- administrar carnitina em todas as AO, e carnitina + glicina nas formas graves de AIV;
- descontaminação intestinal com metronidazol (20 mg/kg) para reduzir a produção de propionato pelo microbioma intestinal (AP, AMM);
- administrar vit B12 (AMM) e biotina (AP);
- promover a remoção dos metabólitos tóxicos com métodos dialíticos: HD (hemodiálise), HF (hemofiltração) ou HDF (hemodiafiltração); a DP (diálise peritoneal) é menos eficaz, especialmente nas situações muito graves, com hiperamoniémia grave (frequente na AP).
Na AMM, como a clearance renal do ácido metilmalónico é alta, se a amoniémia não for muito elevada, poderá ser suficiente a reidratação, a promoção do anabolismo e a dieta hipoproteica; - benzoato de sódio e carbamilglutamato em casos de amónia elevada; se o lactato estiver muito elevado (o que poderá ocorrer na AP) por deficiência de tiamina, deve esta ser suplementada.
Na fase de manutenção
- dieta hipoproteica, por vezes suplementada com Mx de AA isenta dos AA cujo metabolismo está afectado;
- administar sempre carnitina, e carnitina + glicina (AIV);
- descontaminação intestinal intermitente (com metronidazol) durante 10 dias em cada mês, ou contínua com metronidazol (1 mês), trimetroprim (1 mês), amoxicilina (1 mês) e assim sucessivamente (na AP e AMM);
- administar hidroxicobalamina (AMM) e biotina (AP) nas formas sensíveis;
- por vezes é necessário recorrer ao TH (AP), ou TH e/ou TR na AMM;
- vigilância dos desequilíbrios dos AA plasmáticos, especialmente dos AA essenciais, e do nível dos AG essenciais, podendo ser necessário suplemento de ácido docosa-hexanóico;
- alimentação entérica nocturna sistemática, nos mais jovens (qualquer que seja o apetite); por vezes, devido às dificuldades alimentares, tão comuns nas AO, há que recorrer à alimentação por sonda nasogástrica ou gastrostomia.
Grupo II
Uma forma especial de AMM pelas suas características e tratamento particulares é a acidúria metilmalónica associada a homocistinúria. Trata-se de um defeito da síntese intracelular da adenosilcobalamina e da metilcobalamina, cofactores, respectivamente, da metilmalonil-CoA mutase e da metionina sintetase.
Manifestações clínicas
As manifestações clínicas podem iniciar-se in utero: restrição de crescimento fetal, dismorfias moderadas, microcefalia e cardiomiopatia dilatada fatal.
Após o nascimento são descritas duas formas:
Forma infantil: de início precoce, mais frequente e grave, multissistémica, progressiva.
Como alterações mais típicas, citam-se: restrição do crescimento, microcefalia, dificuldades alimentares e de ganho ponderal, hipotonia, hidrocefalia, deterioração neurológica, anemia megaloblástica, alterações maculares e síndroma hemolítica urémica.
Forma tardia: sintomatologia mais evidente após os 4 anos de idade – regressão neurológica, sintomas neuropsiquiátricos, encefalopatia progressiva, degenerescência da espinhal medula, anemia megaloblástica, complicações tromboembólicas.
Diagnóstico
O diagnóstico desta forma especial depende dos resultados dum conjunto de exames laboratoriais: – cromatografia de AA plasmáticos (CAA); – cromatografia de ácidos orgânicos urinários (CAO); – doseamento do ácido metilmalónico e de homocisteína total (tHcy) no plasma; e perfil das acilcarnitinas no plasma.
O perfil bioquímico desta forma clínica inclui: ácido metilmalónico e homocisteína elevados, cistationina e metionina baixos; os níveis de vitamina B12 dentro do normal e de acilcarnitina C3 elevados constituem o padrão clássico.
Tratamento
O tratamento inclui administração diária parentérica de hidroxicobalamina (não a cianocobalamina) parentérica, betaína, ácido folínico e carnitina oral. Está em estudo o uso de hidroxicobalamina intranasal; a administração por via oral é ineficaz.
A dieta hipoproteica é controversa, porquanto as proteínas dietéticas têm muito pouca homocisteína. Podendo ser necessários suplementos de metionina, não deve administrar-se Mx de AA sem metionina (usadas na AMM) pelo risco de hipometioninémia grave.
Estão ainda indicadas:
- dose antiagregante plaquetária de ácido acetilsalicílico;
- monitorização regular da concentração de tHcy no plasma.
Como medida pré-natal importante cita-se a necessidade de reduzir as concentrações dos metabólitos tóxicos na mãe, o que parece ter impacte nas complicações de longo prazo.
Complicações
As complicações são variadas por atingirem diversos sistemas: SNC, olhos, sangue, vasos sanguíneos, rim, coração. A melhor estratégia para as evitar e ou conter diz respeito à administração de doses correctas diárias de hidroxicobalamina parentérica + betaína.
Na forma infantil pode ocorrer perda progressiva de visão até à cegueira (primeira década de vida). As complicações vasculares determinam maior morbilidade e mortalidade.
Salienta-se que no contexto de analgesia/ anestesia/ cirurgia não deve ser usado o óxido nitroso por ser potencialmente tóxico nesta doença.
Recomenda-se, se possível, o uso limitado das Mx de AA específicas, o que poderá facilitar o ajuste individualizado do suprimento de proteínas naturais. O doseamento da ureia e da creatinina na urina de 24 horas será de grande utilidade para esse cálculo. Assim, se o doente estiver medicado com Mx de AA, a razão ureia/ creatinina urinária será elevada, geralmente > 30, o que significa que muitos dos aminoácidos da Mx são excretados pela urina, sem qualquer efeito; se a ureia urinária (que reflecte o catabolismo proteico) for muito baixa e o doente não estiver a tomar a Mx de AA, pode-se, com alguma segurança, aumentar o suprimento proteico, fundamental para o equilíbrio metabólico, nutricional e o crescimento. Estão em estudo:
Abreviaturas: Mx <> misturas |
7. DOENÇAS DO CICLO DA UREIA
ETIOPATOGÉNESE
O ciclo da ureia ou de Krebs-Henseleit que, na sua forma completa, tem lugar somente no fígado, constitui a principal via metabólica comum para a excreção do azoto. A sequência de reacções que o integram, em parte na mitocôndria, em parte no citosol, converte (sobretudo a partir da glutamina e do glutamato) a amónia tóxica e outros compostos nitrogenados em produto não tóxico – a ureia – excretada através da urina.
Aos diferentes defeitos genéticos responsáveis por deficiência de uma ou mais enzimas que intervêm no ciclo (em número de oito – Figura 2) – correspondem diversas entidades clínicas adiante discriminadas em que se verifica hiperamoniémia. De referir, no entanto, que defeitos enzimáticos noutras vias metabólicas poderão secundariamente bloquear qualquer passo do ciclo da ureia.

FIGURA 2. Ciclo da ureia e vias alternativas de excreção de azoto. 1) Carbamil fosfato sintetase; 2) Ornitina transcarbamilase; 3) Arginino-succinato sintetase; 4) Arginino-succinato liase; 5) Arginase; 6) N-acetilglutamato sintetase; 7) Glutamina sintetase; 8) Citrina (transportador mitocondrial de aspartato-glutamato).
As vias alternativas de excreção do azoto, nomeadamente a conjugação da glicina com o benzoato, e da glutamina com o fenilacetato poderão ser aproveitadas como meio de tratamento dos doentes com défice de formação de ureia e hiperamoniémia.
Aspectos epidemiológicos
As doenças do ciclo da ureia (DCU) correspondem aos erros metabólicos hereditários dos mais frequentes (incidência cumulativa de 1/8.000).
Descrevem-se as seguintes entidades clínicas:
- deficiência de ornitina transcarbamilase (OTC);
- deficiência de arginino-succinato sintetase (AS) ou citrulinémia (vários tipos);
- deficiência de arginino-succinato liase (AL);
- deficiência de arginase ou hiperargininémia;
- deficiência de glutamina-sintetase (GS);
- dficiência de carbamil fosfato sintetase (CPS);
- deficiência de N-acetilglutamato sintetase (NAGS); e
- defeito da citrina.
Com excepção da deficiência da ornitina transcarbamilase ou OTC (de transmissão hereditária ligada ao cromossoma X e a forma mais comum de todas as doenças do ciclo da ureia), os outros defeitos são de transmissão autossómica recessiva. Como regra geral, os homozigotos com OTC do sexo masculino têm formas mais graves que os heterozigotos do sexo feminino; por outro lado, os heterozigotos do sexo feminino podem ter formas ligeiras, sendo que cerca de 75% são assintomáticos.
Manifestações clínicas e laboratoriais
As manifestações clínicas das doenças do ciclo da ureia são extremamente variáveis:
No RN aparentemente saudável com peso adequado à idade, após um intervalo livre por vezes inferior a 24 horas, ou de alguns dias, surge anorexia, recusa alimentar, vómitos, letargia, e/ou irritabilidade e taquipneia. Verifica-se deterioração rápida com alterações neurológicas, alterações do tono muscular, hiporreflexia, instabilidade vasomotora, hipotermia, apneia e convulsões, podendo seguir-se coma profundo e morte.
As complicações são: hemorragia cerebral e pulmonar; e, como sequela: grave atraso do neurodesenvolvimento. O diagnóstico inicial sugere habitualmente septicémia, sendo que a presença de alcalose respiratória, associada às manifestações descritas, poderá ser a chave para o diagnóstico.
A ureia plasmática muito baixa (1-2 mg/dL) é um dado relevante; portanto:
|
Após o período neonatal, as manifestações poderão ser menos agudas e mais variáveis: anorexia, letargia, vómitos, hepatomegália, má progressão ponderal, atraso do neurodesenvolvimento, episódios de irritabilidade; diplegia, tetraplegia espástica na deficiência da argininase (argininémia); alterações do cabelo (tricorexis nodosa) na deficiência da arginino-succinato liase.
No adolescente e no adulto: habitualmente verificam-se sintomas neurológicos e/ou psiquiátricos crónicos, com alterações do comportamento, por vezes bizarro, com desorientação, letargia, alterações do estado de consciência, e quadro de psicose. As referidas doenças poderão também manifestar-se por encefalopatia recorrente, geralmente associada a ingestão de elevado teor de proteínas, infecção, estresse, anestesia, estado catabólico ou, por vezes, sem causa aparente.
Na hiperargininémia, o quadro poderá ser diferente, caracterizando-se fundamentalmente por diplegia espástica, (por vezes interpretada como fazendo parte de paralisia cerebral), hiperactividade, ataxia, atetose, distonia e, raramente, coma e convulsões refractárias ao tratamento anticonvulsante.
Diagnóstico
O Quadro 1 mostra situações que, não sendo doenças do ciclo da ureia, podem cursar com hiperamoniémia.
QUADRO 1 – Situações que apresentam hiperamoniémia.
|
Salienta-se que, das hiperamoniémias não devidas a artefactos, cerca de 2/3 correspondem efectivamente a DCU.
No que respeita ao diagnóstico das doenças do ciclo da ureia, é importante rever algumas definições, dando ênfase aos exames analíticos.
Assim, fala-se de hiperamoniémia quando o valor da amónia no sangue é > 80 µmol/L no RN, e > 50 µmol/L após os 28 dias de vida. Na prática, consideram-se valores normais, respectivamente os valores < 50 µmol/L no RN, e < 35 µmol/L após o período neonatal.
Num RN sem doença deste foro, mas com patologia relacionada com septicémia ou asfixia, raramente a amónia é > 180 µmol/L; se o valor for > 200 µmol/L há que suspeitar de doença metabólica, sendo que nas doenças do ciclo da ureia, e designadamente neste grupo etário, são atingidos valores de amónia > 1.500 µmol/L, sem cetoacidose e, geralmente, sem hipoglicémia.
Salienta-se, a propósito, que a hiperamoniémia é uma emergência médica.
Nas situações de suspeita clínica de doença do ciclo da ureia torna-se fundamental realizar de imediato um conjunto de análises básicas obedecendo a rigorosas condições técnicas de colheita e transporte (designadamente amostras de sangue e urina em recipientes acondicionados em gelo); no sangue – amónia, electrólitos, pH e gases, hiato iónico, lactato, glucose, ureia, creatinina, provas de função hepática e factores de coagulação; na urina – análise sumária.
Como análises especiais (em laboratório especializado) estão indicados os seguintes doseamentos: aminoácidos plasmáticos e urinários, ácidos orgânicos urinários, ácido orótico na urina, e perfil da carnitina e acilcarnitinas no plasma. Na deficiência de OTC verifica-se: aumento importante de ácido orótico na urina. Em todas as DCU há acumulação de glutamina (excepto na deficiência de glutamina sintetase/GS) e de alanina.
Os resultados obtidos quanto ao padrão de aminoácidos plasmáticos poderão ter valor diagnóstico para as entidades clínicas a seguir discriminadas: deficiência de arginino-succinato-sintetase (AS), de arginino-succinato liase (AL), de arginase, e de glutamina-sintetase (GS).
O diagnóstico pode ser confirmado, quer por estudo enzimático (leucócitos, fibroblastos, hepatócitos), quer por análise mutacional.
Salienta-se que o doseamento urgente da amónia no sangue deve fazer parte da investigação básica obrigatória em todos os doentes com encefalopatia não esclarecida, em qualquer idade. |
Reiterando que a hiperamoniémia implica tratamento emergente, há no entanto que atender a uma eventual situação de coma hiperamoniémico com duração superior a 2-3 dias: a equipa médica responsável deverá discutir com os pais do paciente as opções a tomar.
Tratamento
O tratamento de emergência compreende:
- interrupção imediata do suprimento proteico durante 24-48 horas;
- aplicação imediata de sonda cânula ou cateter IV para suprimento energético elevado à base de soluto de glucose (a 10% se em veia periférica ou a 10-25% se em veia central); contudo, está indicada restrição de fluidos se houver suspeita de edema cerebral;
- se os vómitos forem persistentes, pode usar-se ondansetron IV na dose de 0,15 mg/kg em 15 minutos, podendo repetir-se até 3 doses diárias;
- introdução imediata de drogas eliminadoras de amónia:
*benzoato de sódio até 500 mg/kg/dia, PO ou IV em 2 doses (1ª de 250 mg/kg em 2-4 horas; 2ª de 250 mg/kg nas próximas 20-22 horas);
*fenilbutirato de sódio até 600 mg/kg/dia, PO ou IV em 2 doses (1ª de 250 mg/kg em 2-4 horas; 2ª de 350 mg/kg nas próximas 20-22 horas);
*L-arginina PO ou IV (até 700 mg/kg/dia na citrulinémia e acidúria arginino-succínica); até 150 mg/kg/dia nas deficiências de ornitina transcarbamilase/ OTC, de carbamilfosfato-sintetase/CPS e de N-acetilglutamato-sintetase/ NAGS; também se usa o carbamil-glutamato nos defeitos de NAGS e CPS, o qual é activador da CPS, primeira enzima do ciclo da ureia – dose inicial de 100 mg/kg/dia, ulteriormente até 300 mg/kg/dia;
*L-carnitina PO ou IV na dose de 200 mg/kg/dia, se o doente estiver submetido a tratamento com benzoato de sódio e emulsão de lípidos (AP); - técnicas de diálise; se a amoniémia for muito elevada (> 400 µmol/L), o doente estiver em coma, a amoniémia não descer significativamente nas primeiras 4 horas do tratamento atrás indicado, ou houver falência multiorgânica, estão indicadas técnicas de diálise: HD (hemodiálise), HF (hemofiltração) ou HDF (hemodiafiltração); em alternativa, diálise peritoneal, menos eficaz; não se deve fazer exsanguinotransfusão; o recurso à ECMO é muito eficaz;
- introdução cautelosa da dieta de emergência sem proteínas nas primeiras 24-48 horas através de alimentação entérica (AE) e/ou alimentação parentérica (AP) consoante a gravidade da encefalopatia aguda, e intolerância digestiva;
- após 24-48 horas sem proteínas, início de suprimento de proteínas- 0,5 g/kg/dia (na AP, sob a forma de aminoácidos) ou na AE através de fórmula infantil adequada à restrição proteica;
- se surgirem convulsões, não usar valproato nem corticóides; os riscos nesta fase são hiperidratação, edema cerebral e má-nutrição;
- se for necessário proceder a transfusão, usar apenas sangue fresco;
- evitar a toxicidade de drogas; sendo impossível o respectivo doseamento sérico, utilizar o valor do hiato iónico: se > 15 mEq/L ou incremento de 6 mEq/L em relação a valor anterior, é provável o estado de toxicidade.
O tratamento de manutenção baseia-se em:
- dieta hipoproteica com restrição de proteína natural de acordo com o tipo de doença, idade, peso, e tolerância individual, suplementada com mistura apropriada de aminoácidos;
- administração de fármacos eliminadores de amónia, per os, como o fenilbutirato de sódio e/ou benzoato de sódio, cloridrato de arginina (excepto no defeito da arginase); na deficiência de arginina-succinato-liase (AL) a dose de arginina deverá ser muito mais baixa do que a usada na fase aguda (manutenção da arginina entre 50-200 µmol/L;
- suprimento adequado de vitaminas, minerais e oligoelementos;
- os fármacos devem ser administrados durante as principais refeições para rendibilizar a remoção da amónia.
Os objectivos do tratamento são:
- manter amoniémia < 80 µmol/L, e glutamina < 800-1.000 µmol/L;
- manter ausência de ácido orótico na urina;
- manter a normalidade dos níveis plasmáticos de proteínas totais, de albumina e pré-albumina, de aminoácidos essenciais (manter Isol > 25 µmol/L – valor baixo é marcador de má-nutrição proteica) e de carnitina total (> 30 µmol/L).
BIBLIOGRAFIA
Abily-Donval L, Dupic L, Joffre C, et al. Management of 35 critically ill hyperammonemic neonates: Role of early administration of metabolite scavengers and continuous hemodialysis. Arch Pediatr 2020;27:250-256
Andrade F, Villate O, Couce ML, et al. Asymmetric dimethylarginine as a potential biomarker for management and follow-up of phenylketonuria. Eur J Pediatr 2019;178:903-911
Bao P-Z, Ye J, Han L-S, et al. Application of isoxanthopterin as a new pterin marker in the differential diagnosis of hyperphenylalaninemia. World J Pediatrics 2019;15:66-71
Blau N, Koch R, Matalon R, e tal. New development on phenylketonuria and tetrahydrobiopterin research. Mol Genet Metab (Suppl 1) 2005;86:1-156
Blau N, Scriver Cr. New approaches to treat PKU: how far are we? Mol Genet Metab 2004;81:1-2
Brusilow SW. Hyperammonemic encephalopathy. Medicine (Baltimore) 2002;81:240-249
Cabral A, Tasso T, Eusébio F, Gaspar A. Novo tratamento da fenilcetonúria em adolescentes e adultos. Acta Pediatr Port 2003;34:271-276
Cabral A, Portela R, Tasso T, Eusébio F, et al. Tratamento de crianças fenilcetonúricas: 27 anos de experiência no serviço de Pediatria do HSM. Rev Port Pediatr 1993;24:55-59
Chace D, Kalas T. A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem 2005;38:296-309
Cleary MA, Skeath R. Phenylketonuria. Pediatr Child Health 2019;29:111-115
Feldmann R, Schallert M, Nguyen T, et al. Children and adolescents with phenylketonuria display fluctuations in their blood phenylalanine levels. Acta Paediatr 2019;108:541-551
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Koch R. Maternal phenylketonuria : the importance of early control during pregnancy. Arch Dis Child 2005;90:114-115
Lyon G, Kolodny EH, Pastores GM (eds). Neurology of Hereditary Metabolic Diseases of Children. New York: McGraw-Hill, 2006
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Nagasaka H, Komatsu H, Ohura T, et al. Nitric oxide synthesis in ornithine transcarbamylase deficiency: possible involvement of low no synthesis in clinical manifestations of urea cycle deficiency. J Pediatr 2004;145:259-262
Sun W-H, Wu B-B, Wang Y-O, et al. Identification of eight novel mutations in 11 Chinese patients with maple syrup urine disease.World J Pediatr 2020;16:401-410
Techakittiroj C, Cunningham A, Hooper PF, et al. High protein diet mimics hypertyrosinemia in newborn infants. J Pediatr 2005;146:281-282
Vega MW, Swartz SJ, Devaraj S, et al. Elevated serum creatinine: but is it renal failure? Pediatrics 2020, 146 (1) e20192828
Wilcken B. Problems in the management of urea cycle disorders. Mol Genet Metab 2004;81:S86-S91