Nomenclatura e importância do problema
Os fungos são seres vivos eucarióticos (estruturas somáticas filamentosas) que, por definição, e ao contrário das bactérias, possuem parede celular composta de quitina, núcleo e organelos (ou organitos) intracelulares.*
A classificação dos fungos não é um assunto fácil. Entre os estudiosos e especialistas existem diferentes opiniões, o que traduz que ainda muito se desconhece sobre a estrutura e fisiologia destes seres vivos. Assim, a interpretação que cada um faz conduz a uma falta de uniformidade nos critérios de definição de determinadas espécies fúngicas.
De acordo com literatura científica, os fungos integram o reino Eumycota; os fungos, também designados Eumycetes (Eumicetas ou fungos verdadeiros), dividem-se em três classes de acordo com o tipo de reprodução sexuada: antigo Zycomycetes (actual Mucorales e Entomophthorales), Ascomycetes e Basidiomycetes. A classificação dos fungos tem sido modificada pela aplicação de novas técnicas moleculares e de espectrofotometria. De facto, hoje em dia a taxonomia baseia-se não só no modo de reprodução sexuada, mas também na concordância das sequências de DNA de zonas hipervariáveis. Surgiu assim a noção de espécies morfologicamente semelhantes, distintas apenas geneticamente e com padrões distintos de susceptibilidade aos antifúngicos, as espécies crípticas, com implicações clínicas importantes.
*Células eucarióticas são aquelas em que há uma divisão nítida entre o núcleo (com membrana nuclear e mais que 1 cromossoma) e o citoplasma (com organitos específicos, por ex. mitocôndrias). Estas células variam muito em tamanho [por ex. diâmetro entre 6-10 um (levedura pequena), até 2 mm de diâmetro e 3-5 cm de comprimento (por ex. a alga unicelular Acetabularia)]. Células procarióticas, são aquelas em que não há divisão entre núcleo e citoplasma; são de menores dimensões e menos complexas estrutural e funcionalmente que as eucarióticas – por ex. são do tamanho dos organitos destas – As bactérias são células procarióticas. |
De uma forma geral, de acordo com a morfologia e tipo de crescimento, os fungos podem ser classificados em 3 tipos:
- Leveduriformes, unicelulares, reproduzindo-se por gemulação, processo pelo qual a célula mãe origina outra, idêntica a si própria (por ex. Cryptococcus);
- Filamentosos, multicelulares, sendo designados por hifas os filamentos longos, os quais, no conjunto formam um micélio; os tecidos são parasitados como filamentos, ou como filamentos e esporos;
- Dimorfos; possuem características completamente diferentes in vivo e in vitro; nos tecidos revelam-se como células leveduriformes (Histoplasma, Blastomyces, Sporotrichum), esferas cheias de endosporos (Coccidioides imitis) ou células fumagóides (agentes de cromomicose); e em meios de cultura correntes e a 24ºC, originam colónias
Das particularidades biológicas poderá resultar alguma confusão dos nomes atribuídos às diferentes formas; por ex., à forma unicelular de determinado fungo dá-se o nome de Cryptococcus neoformans, e de Filobasidiella neoformans à forma hifa. O agente Pneumocystis jiroveci (ex- carinii), anteriormente considerado protozoário, é hoje englobado na categoria dos fungos.
Das cerca de 100.000 espécies de fungos existentes, apenas uma pequena minoria é patogénica para o ser humano, designando-se por micoses as infecções causadas por fungos.
Numa perspectiva simples, de prática clínica, uma das classificações considera os seguintes tipos, de acordo com a localização da infecção: 1 – superficiais (cutaneomucosas); 2 – subcutâneas; 3 – sistémicas ou profundas.
Na criança saudável, as infecções sistémicas graves por fungos são pouco frequentes. Nas últimas duas décadas, tem-se verificado um aumento de infecções fúngicas graves em crianças imunodeprimidas, o que pode estar relacionado com estratégias terapêuticas cada vez mais agressivas, podendo levar a neutropénia prolongada.
Com efeito, cerca de 8% a 10% dos episódios febris em doentes imunocomprometidos são devidos a infecções invasivas por fungos. Os agentes etiológicos mais frequentes nestas situações são Candida albicans, Aspergillus fumigatus e Cryptococcus neoformans.
Contudo, mais recentemente, em parte devido à utilização profiláctica de fármacos antifúngicos, têm emergido outros fungos como causa de doença invasiva.
Etiopatogénese
Estão descritos três mecanismos principais pelos quais é adquirida a doença infecciosa por fungos:
- Inoculação/contaminação; em geral, o fungo afecta o hospedeiro imunocompetente, sendo a infecção resultante da contaminação cutânea ou inoculação, como por exemplo as dermatofitoses ou dermatofitias (por fungos filamentosos com afinidade específica para as estruturas ceratinizadas) e as candidíases mucocutâneas;
- Doença sistémica (primária); nesta categoria estão incluídas as infecções por fungos com virulência suficiente para infectar um hospedeiro imunocompetente. São exemplos: histoplasmose, coccidioidomicose, blastomicose e paracoccidioidomicose;
- Doença/micose oportunista; neste tipo de mecanismo estão englobados agentes com menor virulência que raramente causam doença invasiva no hospedeiro imunocompetente. São exemplos: candidíase invasiva, aspergilose e criptococose.
Neste capítulo é dada ênfase às infecções fúngicas superficiais e sistémicas.
As infecções subcutâneas: a – Esporotricose (por Sporothrix schenckii); b – Micetomas (por Madurella micetomi e outras espécies); c – Cromomicose (por Fonsecae pedrosoi); d – Rinosporidiose (por Rinosporidium seeberi) são habitualmente do domínio do Dermatologista.
1. INFECÇÕES FÚNGICAS SUPERFICIAIS
1.1 Dermatofitoses mais comuns
Os fungos dermatófitos alojam-se na camada superficial da epiderme, unhas e cabelo, onde proliferam. Não invadem as camadas inferiores da epiderme ou derme. Os agentes mais frequentemente implicados são Trichophyton, Microsporum e Epidermophyton.
No que respeita à epidemiologia, verifica-se distribuição mundial. A infecção é adquirida por contacto directo com humanos ou animais infectados ou, no caso de dermatófitos geofílicos, por contacto com o solo.
Nesta alínea são abordadas as dermatofitias mais frequentes, a pitiríase versicolor e as candidíases mucocutâneas.
Tinea capitis
Esta dermatofitia é causada por fungos do género Trichophyton e Microsporum, tais como M. canis, M. audouinii, M. mentagrophytes e T. tonsurans (90% dos casos nos EUA). Os agentes causais variam consoante a área geográfica.
É muito frequente em idade pediátrica, sobretudo em crianças com 3-7 anos. As crianças e os adultos podem ser portadores assintomáticos. A incidência é maior em afro-americanos, condições de higiene deficitárias e baixo nível socioeconómico.
Pode atingir o couro cabeludo, sobrancelhas e pestanas, sendo a apresentação clínica variável: dermatose descamativa não inflamatória, inflamação com lesões eritematosas e descamativas acompanhadas de alopécia, podendo progredir para lesões mais acentuadas, tipo foliculite (kerion); pode manifestar-se igualmente por nódulos supurativos ou por lesões tipo favo (escútula fávica/tinha favosa, com crostas e escamas amareladas e aderentes); por vezes acompanha-se de febre e adenopatias satélites. Após cicatrização, pode haver alopécia definitiva. A evolução depende da interacção agente-hospedeiro. (Figura 1)
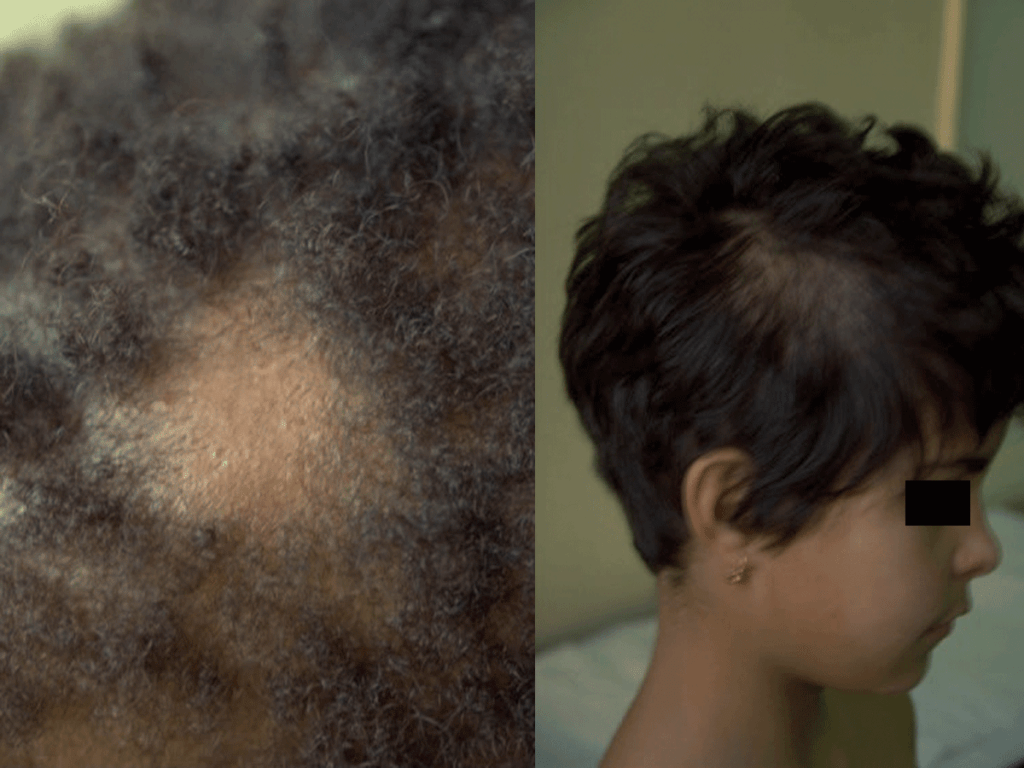
FIGURA 1. Tinea capitis. (NIHDE)
O diagnóstico é clínico, mas deve ser confirmado através da observação do fungo por microscopia óptica, sobretudo na tinha do couro cabeludo. O exame cultural é o meio de identificação do agente específico.
O tratamento é feito com griseofulvina PO, 10-20 mg/kg/dia, em 1 dose diária, durante 6 a 12 semanas; terbinafina (< 20 Kg, 62,5 mg/dia; 20-40 kg, 125 mg/kg; > 40 kg, 250 mg/dia), 2 a 6 semanas, itraconazol 3 a 5 mg/kg/dia 2 a 4 semanas ou fluconazol 6 mg/kg/dia 2 a 4 semanas. Os champôs com sulfureto de selénio a 2,5% ou à base de cetoconazol a 2% reduzem a propagação da infecção.
Tinea unguium
A tinha das unhas deriva da invasão das lâminas ungueais por dermatófitos, sendo que a designação de onicomicose é mais lata do que a de tinha das unhas; com efeito, aquela inclui toda a distrofia ungueal causada por qualquer espécie de fungos, sejam dermatófitos, Candida, ou outros.
Os agentes mais frequentes da tinha das unhas são T. rubrum e T. interdigitale.
As manifestações clínicas variam entre pequenas manchas esbranquiçadas até espessamento com destruição da lâmina da unha e hiperceratose subungueal. Distinguem-se dois tipos clínicos: o distal, mais frequente, e o proximal, iniciado na prega peri-ungueal, em regra perto da lâmina. Observa-se discromia (cor esbranquiçada ou amarelada), superfície irregular, por vezes baça, fendilhação e descolagem que chega a separar a unha em duas lâminas. A onicólise total é rara. A doença atinge uma ou várias unhas. É raro que todas estejam alteradas, o que constitui elemento de diagnóstico em relação a outras afecções como a psoríase.
O diagnóstico faz-se através da identificação do agente por microscopia óptica e exame cultural. O tratamento da tinha das unhas é feito com griseofulvina, durante 6 a 12 meses ou terbinafina 6-12 semanas. Outras opções terapêuticas são o itraconazol e o fluconazol, de modo contínuo ou cíclico. Geralmente os antifúngicos tópicos são ineficazes pois não atingem as camadas inferiores do leito ungueal, mas poderão ser utilizados em associação com a terapêutica sistémica.
O tratamento da onicomicose por Candida é abordado na alínea 1.2.
Tinea corporis
A tinha do corpo ou tinea corporis atinge as áreas de menor pilosidade e não apenas a verdadeira pele glabra, como a das palmas das mãos e a das plantas dos pés. Pode atingir a face, tronco e extremidades.
Os agentes etiológicos são fungos do género Trichophyton (espécies T. rubrum, T. mentagrophytes, T. tonsurans), Microsporum (M. canis) e Epidermophyton (E. floccosum).
A lesão mais comum é a impigem, pruriginosa. Após período de incubação de 3 a 30 dias, o fungo desenvolve-se dentro e paralelamente à superfície da camada córnea. A lesão inicia-se por algumas vesículas com aspecto herpético. Estas vesículas passam por vezes despercebidas. Atenuam-se e surge em seguida a figura anular/circular designada classicamente como “herpes circinado”, com bordo vesiculoso, mais ou menos inflamatório, centro descamativo, e crescimento centrífugo. Da confluência de várias lesões resulta por vezes um aspecto policíclico. O prurido na zona da impigem é factor de disseminação pela coceira que origina, sobretudo em indivíduos com alteração imunológica local ou geral, congénita ou devida a terapêutica imunossupressora. (Figura 2)
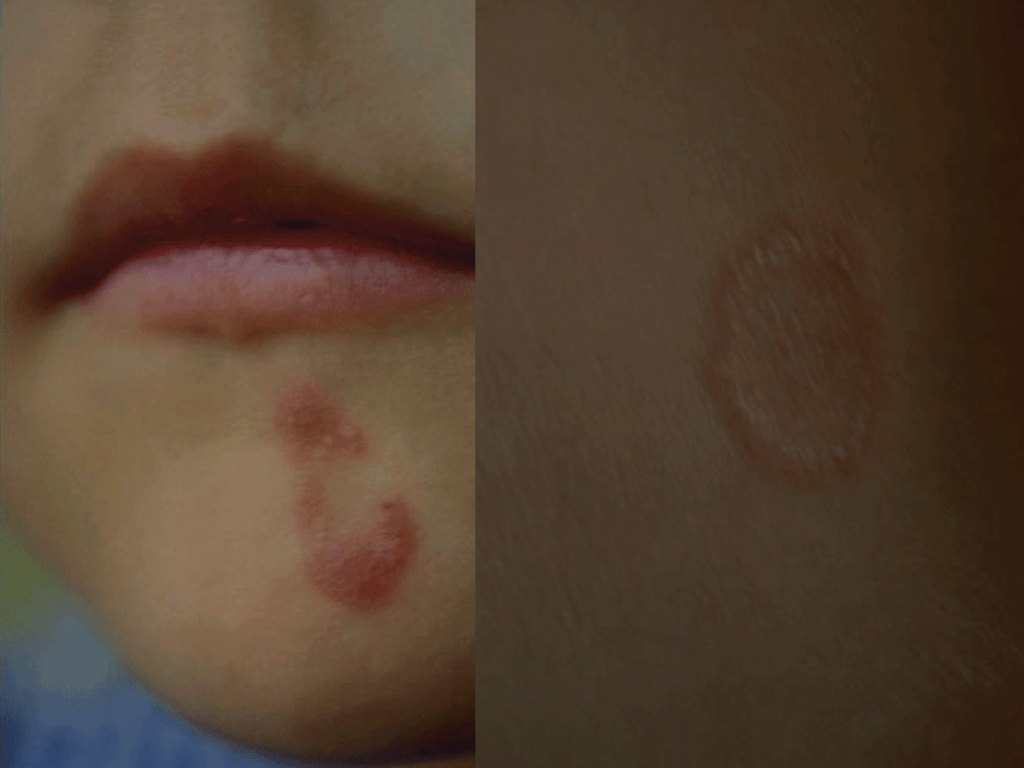
FIGURA 2. Tinea corporis: impigem. (NIHDE)
Tinea pedis (pé de atleta)
As lesões dos pés (tinha dos pés ou “pé de atleta”) são provocadas por fungos do género Epidermophyton (E. floccosum) e Trichophyton (T. rubrum, T. mentagrophytes). Situam-se nos espaços interdigitais, na planta e no bordo. São constituídas por vesículas e pústulas que rebentam, secam e descamam, às quais sucedem maceração, fissuras, hiperceratose e alterações das unhas. A evolução é geralmente cíclica, com exacerbação no tempo quente e tendência para a cronicidade. O quadro clínico pode ser complicado por agentes piogénicos. Por vezes aparece linfangite.
A designação de “pé de atleta” deve o seu nome ao facto de ser muito comum entre desportistas praticando em ginásios, balneários e piscinas, assim como nos casos de uso prolongado de calçado favorável à acumulação de humidade. Contudo, este tipo de micose pode afectar qualquer indivíduo.
Tinea cruris
Certos factores predisponentes (pele fina, obesidade, atrito/roupa apertada, temperatura e humidade), agravados por certos tipos de vestuário, condicionam as características clínicas e a evolução da tinha das virilhas, também chamada tinea cruris.
Esta micose é provocada por fungos do género Epidermophyton (E. floccosum) e Trichophyton (T. rubrum, T. mentagrophytes).
As manifestações clínicas iniciam-se por pápula ou pequena área eritematosa, elevada, a qual invade as pregas das virilhas, períneo, e por vezes nádegas, onde a expressão clínica é a de impigem. O centro das lesões das virilhas é habitualmente castanho-avermelhado. Por vezes verifica-se descamação ou maceração e mesmo fissura; em certos casos há apenas descamação. É mais frequente em rapazes após a puberdade. Pode haver transmissão interpessoal.
O tratamento antifúngico indicado nas situações de tinea corporis, tinea pedis e tinea cruris é tópico (nistatina, terbinafina, clotrimazol ou miconazol) durante 4 semanas; contudo, nas lesões crónicas, por vezes múltiplas e recidivantes de tinea corporis da pele glabra, em que se perde o carácter de impigem e a dermatose passa a assemelhar-se a eczema ou psoríase, ou assume forma difusa granulomatosa (rara) com invasão da profundidade dos tecidos e dos gânglios, torna-se necessário tratamento sistémico e, em casos específicos, incisão e drenagem de lesões supuradas.
Pitiríase versicolor (Tinea versicolor)
Trata-se de uma micose muito comum devida a Malassezia furfur. Tem distribuição mundial, mas aparece com maior frequência nas regiões tropicais e subtropicais, e afecta sobretudo adolescentes e adultos jovens; pode verificar-se transmissão interpessoal quando em fase de descamação.
Como manifestações clínicas, referem-se lesões maculares hipo ou hiperpigmentadas, policíclicas, com descamação furfurácea; a sua patogénese não está esclarecida (a descamação não parece funcionar como filtro solar, sendo que se admite um defeito no transporte dos grânulos de melanina, secundário à infecção). As referidas máculas discrómicas têm dimensões variadas, de contornos nítidos ou difusos, por vezes confluentes; localizam-se sobretudo na metade superior do tronco, braços e região cervical, mais ou menos simetricamente.
Muitas vezes, a doença é detectada poucos dias após a exposição da pele ao sol porque nas áreas da pele afectadas pela micose a pele não se bronzeia. No Inverno, as lesões tomam uma coloração escurecida, castanho-avermelhada. É característica desta micose a cor variável (versicolor) associada à descamação que se torna mais nítida por raspagem com a unha – sinal clássico no diagnóstico diferencial com outras alterações discrómicas da pele.
O diagnóstico é habitualmente fácil. Apoia-se nos factos clínicos, na pesquisa do agente nas escamas por microscopia óptica e no exame da pele com radiação ultravioleta negra – luz de Wood. O exame cultural raramente é necessário.
Está indicado tratamento antifúngico tópico (hipossulfito de sódio a 20%, ou cetoconazol a 1%, ou derivados tópicos do imidazol, entre outros); em função do contexto clínico poderá estar indicada terapêutica com antifúngico oral (cetoconazol na dose de 3 mg/kg/dia durante 10 dias). As recaídas são frequentes.
1.2 Candidíase
A candidíase (ou candidose) é um tipo de micose provocada pelo fungo do género Candida. O habitat natural é o tubo digestivo e génito-urinário, onde vive como comensal. C. albicans encontra-se raramente na pele sã, embora seja habitual em mucosas sem alterações.
Mais de 150 espécies foram descritas, e pelo menos 17 podem causar doença invasiva. Na idade pediátrica a estirpe mais frequente é C. albicans, responsável por cerca de metade (44% a 49%) dos casos, logo seguida por C. parapsilosis (22,2% a 34%) que tem aumentado nos últimos anos. C. glabrata e C. krusei são menos frequentes em pediatria, mas estão associadas a resistência aos triazóis.
Os principais factores de virulência são: – as adesinas, que permitem a adesão e colonização; – a produção de enzimas proteolíticas; – a formação de biofilmes (associados ao aparecimento de resistência aos antifúngicos); e – a capacidade de evolução para as formas invasivas (hifas).
As respostas inata e adaptativa são fundamentais no combate à infecção. A resposta inata inicial do hospedeiro envolve o reconhecimento de padrões moleculares dos microrganismos (PAMP) pelos receptores de reconhecimento de padrões (PRR). Esta resposta é fundamental para desencadear a resposta do hospedeiro, nomeadamente através de neutrófilos, macrófagos e monócitos, impedindo a disseminação da infecção.
Também o sistema adaptativo, em especial as células T, são importantes. Em 2005 foi identificado um subtipo de células TCD4+Th17 cruciais para o controlo de C. albicans ao nível da mucosa. De facto, admite-se que os doentes com candidíase crónica mucocutânea (CMC), seja autossómica dominante (por defeito STAT1), por síndroma de hiperIgE (defeito STAT3) ou por produção de autoanticorpos contra a citocina 17, como na poliendocrinopatia autoimune com candidíase (APECED), têm um defeito nesta via.
Candidíase oral
Também vulgarmente conhecida como “sapinhos”, monilíase oral (Monilia sinónimo de Candida na taxonomia clássica), é a mais frequente de todas as formas de candidíase, sobretudo nos primeiros meses de vida; pode ocorrer entre 2%-5% de RN considerados saudáveis. É muito frequente nos doentes com infecção por VIH, neoplasias e outras imunodeficiências, embora possa ocorrer em crianças saudáveis, sobretudo após corticoterapia inalada e antibioticoterapia sistémica.
Como manifestações clínicas há a referir pequenas pápulas ou placas brancas “leitosas” (assemelhando-se a ”restos de leite”) com base eritematosa, confluentes, muito aderentes, dispersas por toda a mucosa (gengivas, língua, e particularmente na mucosa jugal). Quando removidas deixam a descoberto superfície vermelha e sangrenta.
O tratamento pode ser efectuado com antifúngicos tópicos:
- Nistatina em suspensão oral (100 000 U/mL), PO, 1-2 mL no lactente e 4-6 mL na criança mais velha, 4 vezes/dia, durante 7-10 dias; ou
- Miconazol a 20% em gel, PO, 1-2 mL no lactente; 3 a 5 mL na criança mais velha, 3-4 vezes/dia; a duração do tratamento é 7-10 dias, salientando-se a conveniência de prolongar o tratamento alguns dias após cura clínica para evitar recidivas.
A prevenção implica a execução de algumas medidas de higiene como esterilização de chupetas e biberões; nos lactentes alimentados ao peito, deve ser observada a glândula mamária (mamilo/aréola) no sentido de detectar eventuais sinais de candidíase, a qual deve ser tratada.
Na doença grave a moderada, sobretudo no doente imunodeprimido, pode ser necessário antifúngico oral, habitualmente fluconazol 3 a 6 mg/kg/dia, 7 a 14 dias.
Candidíase perineal
Surge tipicamente na região do períneo. Caracteriza-se por lesões eritematosas, de bordos elevados, por vezes com pápulas, vesículas ou pústulas na região perianal. Pode estar associada a candidíase oral. O tratamento consiste em antifúngico tópico (por ex. clotrimazol a 1%, miconazol, cetoconazol), 2-3 vezes/dia, durante 7-10 dias.
Uma vez que esta situação pode estar associada a dermatite das fraldas, está indicada a aplicação, em alternância, de pasta de óxido de zinco. Como medidas preventivas, e em complemento, cabe salientar: mudança frequente de fraldas (na idade das fraldas) e secagem cuidadosa da pele.
Outras infecções superficiais por Candida
Citam-se, de modo sucinto:
- Glossite (que pode surgir após antibioticoterapia);
- Boqueira (sinónimos: ângulo infeccioso ou perlèche) ou lesões de eritema e pequenas fissuras nas comissuras bucais, havendo por vezes factores predisponentes como atopia, imunodepressão, irritação local com pasta dentífrica, elixir, taninos, etc.. Nestas duas situações (glossite e boqueira) aplicam-se os princípios enunciados para o tratamento da candidíase oral;
- Dermatoses eritematosas maculovesiculares, por vezes papulares ou em placas, em zonas de pele húmida ou tapada (espaços interdigitais, axilas, virilhas);
- Onicomicose (rara, muito difícil de erradicar), a qual pode ser observada em adolescentes manuseando água com frequência. Este quadro poderá associar-se a candidíase mucocutânea.
Candidíase esofágica
Geralmente observada nas crianças imunodeprimidas, é uma doença que pode surgir nas situações de síndroma de imunodeficiência adquirida (SIDA). São factores de risco da citada candidíase a imunossupressão por neoplasia, transplante, imunodeficiência primária, exposição a corticóides e antibioterapia de largo espectro.
Os sintomas mais comuns incluem odinofagia, dor retroesternal ou epigástrica e disfagia. Está frequentemente associada a candidíase da orofaringe. Na endoscopia observam-se placas brancas, eritematosas, com edema, ulceração e por vezes estenose da mucosa esofágica. O diagnóstico histológico é fundamental, evidenciando invasão dos tecidos pelo fungo. O tratamento implica antifúngico sistémico, geralmente fluconazol durante 14 a 21 dias.
2. INFECÇÕES FÚNGICAS SISTÉMICAS
Nesta alínea são descritas algumas formas clínicas de infecção fúngica sistémica, salientando-se (dentro da raridade) a sua ocorrência mais frequente nos doentes em estado grave, em geral hospitalizados, e portadores de imunodeficiência de etiologia diversa.
2.1 Candidíase sistémica ou invasiva
Etiopatogénese e importância do problema
Na idade pediátrica, a incidência de candidíase invasiva tem aumentado nos últimos anos, variando entre 35 a 50/100 000 internamentos. A disseminação surge em 8,3% a 17% dos casos e está associada a uma mortalidade elevada (7,7% a 26%).
Merecem referência como grupos mais afectados: o RN pré-termo MBP e os doentes com infecção por VIH/SIDA, cancro e os transplantados com compromisso do estado imunitário.
Mais recentemente, em crianças com patologia gastrintestinal, nomeadamente síndroma do intestino curto ou submetidos a intervenções cirúrgicas complexas, verifica-se incremento do risco.
São poucos os estudos que definem os factores de risco de doença invasiva por Candida na idade pediátrica. A presença de cateter venoso central, alimentação parentérica, prescrição prévia de vancomicina ou de antibióticos com espectro de acção para anaeróbios, e a terapêutica imunossupressora em doentes transplantados ou com doença oncológica, foram alguns dos factores identificados.
Manifestações clínicas
Os sinais clínicos de candidíase sistémica são os de sépsis por outros agentes. No lactente e RN é mais frequente a meningite, o choque séptico e a candidémia persistente. A candidíase congénita é rara, mas pode surgir no recém-nascido de termo, manifestando-se como eritema difuso e neutrofilia nas primeiras 24 horas de vida. A candidíase disseminada pode afectar meninges, olhos, coração, pulmões, rins ou ossos. É muito frequente no doente oncológico, sobretudo após transplante de medula óssea. Se não tratada, pode evoluir para um quadro de choque séptico (forma aguda) ou para candidíase crónica disseminada (forma crónica).
A doença renal é manifestada por candidúria; contudo, o isolamento de Candida na urina poderá traduzir eventual colonização, cistite ou pielonefrite com ou sem micetomas (bolas de fungos). Poderão surgir microabcessos, necrose papilar, distorção dos cálices e obstrução da via excretora (do lume ou exterior ao lume).
A meningoencefalite é mais comum no RN, a endocardite surge habitualmente em crianças com valvulopatia e a osteomielite, rara, é mais frequente em lactentes e crianças pequenas. O envolvimento ocular pode surgir no contexto de candidémia e doença disseminada, como endoftalmite/retinopatia.
Diagnóstico
O diagnóstico de candidíase sistémica implica o isolamento de Candida de locais habitualmente estéreis. A hemocultura é o exame de eleição, com uma sensibilidade que se aproxima dos 50%. No caso de infecção disseminada, a cultura de tecidos por biópsia poderá ser fundamental para o diagnóstico. O isolamento de Candida nas secreções respiratórias indica colonização e raramente necessita de terapêutica antifúngica.
Nas infecções invasivas é fundamental determinar a susceptibilidade aos triazóis das estirpes isoladas. A detecção de antigénios (como β-D-gulcano) e as técnicas de biologia molecular (PCR) não estão ainda padronizadas para a idade pediátrica.
Em função do contexto clínico, outros exames devem ser realizados:
- Exame oftalmológico para detecção de retinite ou outras alterações do sistema ocular; aliás, na infecção fúngica sistémica este exame torna-se obrigatório;
- Hemoculturas seriadas até ficarem negativas;
- Na suspeita de meningite deve ser analisado o LCR por punção lombar (em particular no RN), na ausência de contraindicação;
- Sugere-se a avaliação renal, cardíaca e hepática nos doentes imunodeprimidos graves e nos doentes com fungemia persistente;
- Outros exames de imagem (ecografia, TAC, RM, outros) devem ser ponderados em função do território afectado.
Tratamento
Nos últimos anos têm sido emitidas várias recomendações das sociedades americanas e europeias sobre a terapêutica da candidíase sistémica na idade adulta, com algumas indicações para a pediatria. Salienta-se, contudo, que a terapêutica deve ser sempre adaptada à epidemiologia local, nomeadamente ao tipo de doente, à estirpe isolada e à exposição prévia a antifúngicos.
Assim, de um modo geral, perante a suspeita de candidíase sistémica no doente não neutropénico, a terapêutica empírica inclui fluconazol (8 a 12 mg/kg/dia) ou anfotericina B (habitualmente a lipossómica, na dose de 3-5 mg/kg/dia), ou equinocandina (caspofungina 70 mg/m2/dia, seguida por 50 mg/m2/dia ou micafungina 2-4 mg/kg/dia).
Nos doentes hemodinamicamente instáveis, com doença grave, sob terapêutica profiláctica com fluconazol, ou com infecção prévia por C. krusei ou C. Glabrata, deve dar-se preferência a uma equinocandina ou a anfotericina B lipossómica. No doentes com infecção por C. parapsilosis é preferível optar por fluconazol ou anfotericina B lipossómica.
No período neonatal pode ser utilizada a anfotericina B convencional, uma vez que a toxicidade renal é menos frequente. Nos casos de infecção grave do SNC e endocardite deve associar-se à anfotericina B lipossómica a flucitosina (50-100 mg/kg/dia, em 4 doses com doseamento sérico bissemanal).
A terapêutica deve ser iniciada o mais precocemente possível e mantida durante 14 dias após candidémia negativa, desde que se exclua foco não resolvido ou não haja imunodeficiência grave. Havendo cateter venoso central, o mesmo deverá ser removido, se possível.
Nos casos de terapêutica empírica iniciada com equinocandina, com estabilidade hemodinâmica, estirpe isolada susceptível ao fluconazol e hemoculturas repetidas negativas, recomenda-se a transição para fluconazol.
No caso de disseminação, a terapêutica deverá ser prolongada de acordo com a focalização e doença de base.
Prognóstico
O prognóstico depende da focalização e do estado imunitário do doente, com uma mortalidade que varia entre 7,7% a 26% nas séries pediátricas.
Nos RN prematuros as infecções invasivas por Candida estão associadas a atraso no neurodesenvolvimento e elevada mortalidade (35% a 66%).
2.2 Aspergilose
Importância do problema
A aspergilose evidencia uma incidência crescente nos últimos anos, associada ao aumento de doentes com imunossupressão grave. A doença invasiva tem geralmente uma mortalidade elevada, sendo actualmente a causa mais frequente de morte por micose sistémica.
Trata-se duma infecção fúngica de larga distribuição mundial. Estão descritas aproximadamente 185 espécies de Aspergillus, das quais cerca de 19 estão associadas a doença humana, na sua maioria por Aspergillus fumigatus e, em menor grau a A. flavus, A. niger, A. terreus e A. nidulans; a identificação destas espécies é importante por motivos terapêuticos.
Aspergillus spp é um membro dos Eumicetas (fungos verdadeiros), produzindo micélio e esporos assexuados (conídias) que são libertados para a atmosfera, podendo ser encontrados em qualquer local, incluindo o ambiente hospitalar.
Etiopatogénese, manifestações clínicas e diagnóstico
Na maioria dos casos, a doença por Aspergillus afecta o pulmão na medida em que o primeiro evento para que aquela surja é a inalação do fungo. Em muitos casos verifica-se a circunstância de obras de construção civil com libertação de poeiras contaminadas com esporos do fungo.
Os neutrófilos e macrófagos são fundamentais na defesa contra a doença invasiva. Os macrófagos alveolares, a primeira defesa, são responsáveis pela eliminação das conídias inaladas. Numa segunda fase os neutrófilos impedem a infecção invasiva.
Ao abordar o tema relacionado com este fungo, é importante falar nas micotoxinas, uma das quais, a aflatoxina, produzida por algumas espécies de A. flavus, que pode contaminar os cereais e outros alimentos; também é um potente carcinogénio cujo papel na doença humana não está esclarecido.
Salienta-se que o agente Aspergillus é altamente angiotrópico.
A infecção por Aspergillus spp pode manifestar-se como 3 síndromas distintas relacionadas com a imunocompetência do hospedeiro:
- Duas formas de aspergilose não invasiva são observadas em doentes com sistema imune normal ou ligeiramente alterado: aspergiloma pulmonar e aspergilose broncopulmonar alérgica (ABPA); e
- Outra forma, aspergilose invasiva, podendo a invasão ser local ou disseminada, afectando os hospedeiros gravemente imunossuprimidos.(#)
(#)Para além das formas descritas, descrevem-se ainda as chamadas síndromas não invasivas saprofíticas em crianças imunocompetentes, traduzidas essencialmente por colonização com o fungo ao nível do canal auditivo externo (otomicose) e aspergiloma pulmonar. |
No caso da ABPA, existe uma resposta alérgica a Aspergillus spp (adquiridos por inalação), por hipersensibilidade de tipo I e III.
No aspergiloma, como o nome parece indiciar, existe acumulação de fungos (micélios) em forma de bola, ocupando uma cavidade pulmonar pré-existente.
A aspergilose invasiva, a forma de apresentação mais grave, ocorre no contexto de doentes com imunodeficiência grave primária (em particular doença granulomatosa crónica), ou adquirida (sobretudo imunossupressão pós-transplante, corticoterapia prolongada, e neutropénia).
A aspergilose pulmonar invasiva manifesta-se de forma subtil e heterogénea nos doentes imunodeprimidos, com tosse seca, febre, dificuldade respiratória, dor pleurítica e novos infiltrados pulmonares como sinais radiográficos; com a progressão da infecção pode associar-se hemoptise (por vezes maciça) e taquicárdia. Cerca de 2-3 semanas depois pode verificar-se cavitação pulmonar no contexto de imunodeficiência e de estado geral grave símile septicémia bacteriana e eventual disseminação. Ao contrário dos doentes neutropénicos em que a doença é aguda e rapidamente progressiva, nos doentes com doença granulomatosa crónica a apresentação é crónica e insidiosa, com astenia, presença ou não de febre, aumento da velocidade de sedimentação e sinais de pneumonia. Neste grupo particular de doentes não ocorre angioinvasão.
No contexto de imunodeficiência e de estado geral grave surge a possibilidade de formação de focos metastáticos em diversos órgãos [seios perinasais (sinusite recorrente, polipose)], pele (placas eritematosas que evoluem para escaras-ecthyma gangrenosum), globo ocular (retinite, celulite orbitária, etc.), SNC (meningite, enfartes e abcessos cerebrais), osso (osteomielite), e coração (endocardite).
O diagnóstico da infecção, considerada de modo global, implica a documentação histopatológica e com exames culturais.
Na prática clínica, para o diagnóstico da ABPA, considera-se imprescindível a verificação dos seguintes critérios: história de obstrução brônquica, eosinófilos no sangue > 500/mm3, IgE total sérica elevada, detecção de precipitinas para Aspergillus, aumento da IgE específicas para Aspergillus, prova cutânea positiva para Aspergillus, sinais radiográficos evidenciando infiltrados pulmonares e bronquiectasias centrais.
De salientar que o diagnóstico da doença invasiva é difícil. Considera-se este diagnóstico se houver identificação de Aspergillus por microscopia ou cultura em tecidos habitualmente estéreis, obtidos por biópsia (tendo em atenção que, encontrando-se no ambiente o Aspergillus, será preciso demonstrar a sua presença intratecidual. Importa referir que a hemocultura é geralmente negativa, mesmo na forma disseminada.
Tratamento
- Nas síndromas de hipersensibilidade estão indicados corticóides sistémicos: prednisona oral (0,5 a 1 mg/kg/dia durante 14 dias, e depois em dias alternados durante 6 a 8 semanas, com redução progressiva). A remissão clínica e radiológica, assim como a redução dos nível de IgE sérico, determinam a interrupção do tratamento.
Nos doentes que necessitam de uma dose elevada de corticóides recomenda-se a associação de itraconazol 5 mg/kg durante 16 semanas, com monitorização dos níveis séricos.
Dado que a doença se correlaciona com níveis elevados de IgE relacionada com a carga fúngica, está em investigação o emprego de anticorpos anti-IgE (omalizumab) associados a antifúngicos na forma broncopulmonar alérgica. - Em situações específicas de aspergiloma está indicada ressecção cirúrgica associada a antifúngicos, ponderados os riscos.
- Na aspergilose invasiva, o voriconazol (PO ou IV) é considerado o agente de primeira linha:
- IV: dose de carga 9-14 mg/kg/dose, seguido de 8 mg/kg de 12/12h; nos adolescentes a dose será semelhante aos adultos (dose de carga 6 mg/kg/dose de 12/12h, seguidas de 4 mg/kg/dose de 12/12h); oral: 9 mg/kg/dose 12/12h (acima dos 50 kg, dose de adulto 200 mg 12/12h).
Este tratamento requer monitorização do nível sérico “em vale” com valores superiores a 1,0 μg/mL). Como agentes alternativos de segunda linha emprega-se a anfotericina B lipossómica (3-5 mg/kg/dia), posaconazol, caspofungina ou micafungina.
A duração do tratamento depende da gravidade da imunossupressão, da localização da doença e da evolução clínica; o mesmo pode ser iniciado IV, passando a PO à medida que se verifica a melhoria.
Prognóstico
O prognóstico é reservado nas formas invasivas, comportando mortalidade ~ 70%; o mesmo está também condicionado pela doença de base.
Prevenção
Nos doentes de alto risco (com cancro, neutropénia e submetidos a quimioterapia) tem sido preconizada a administração de anfotericina B em aerossol, ou de itraconazol PO (2,5-5 mg/kg/dia em duas doses diárias) como quimioprofilaxia. Tendo em conta a etiopatogénese, tais doentes deverão ser afastados de zonas de obras e de poluição.
2.3 Criptococose
Importância do problema
A criptococose é uma doença fúngica invasiva provocada por C. neoformans, uma levedura monomórfica capsulada. Cursa habitualmente com meningoencefalite e pneumonia e afecta tanto doentes imunodeprimidos como imunocompetentes, sendo menos frequente em crianças.
O agente, de distribuição mundial, tem 4 serótipos (A, B, C, D): os serótipos B e C, antes conhecidos por C. neoformans var. gatti e actualmente designados por C. gattii, relacionam-se com infecções prevalentes nas regiões tropicais e subtropicais, designadamente em certas regiões da Austrália e afectam mais frequentemente imunocompetentes; os serótipos A (C. neoformans var. grubii) e D (C. neoformans var. neoformans) são geralmente considerados agentes oportunistas e estão associados a infecções em doentes com imunodeficiência congénita, ou adquirida, sobretudo em relação com infecção por VIH/SIDA e terapêutica imunossupressora em doenças linfoproliferativas.
Etiopatogénese
Admite-se que, na maior parte dos casos, a infecção seja adquirida por inalação de esporos, não se verificando transmissão de pessoa a pessoa. Raramente a infecção pode ser adquirida por via cutânea ou ocular.
As estirpes virulentas de C. neoformans possuem uma cápsula espessa constituída por polissacáridos, que protege o agente infeccioso da fagocitose por macrófagos e neutrófilos. A imunidade celular T é fundamental na defesa contra a infecção, sendo a resposta Th1 protectora e a resposta Th2 facilitadora da infecção. Atingidos os pulmões, pode surgir um quadro de pneumonia com formação de granulomas contendo leveduras, em geral de localização subpleural. Nas circunstâncias de falência do sistema imune para conter a infecção, o agente ultrapassa a barreira alvéolo-capilar e atinge, por via hematogénica, outros órgãos e sistemas (meninges, cérebro, pele, globo ocular, próstata e sistema esquelético). Ou seja, a infecção disseminada pode ocorrer secundariamente a doença pulmonar em doentes com disfunção imunitária de células T, incluindo crianças com leucémia ou linfomas, submetidas a transplante de órgãos sólidos ou com imunodeficiência congénita ou adquirida (SIDA).
Manifestações clínicas
As principais manifestações são:
- Meningite subaguda ou crónica com cefaleias intensas associadas a alterações inespecíficas como febre, astenia, náuseas e vómitos. Podem surgir alterações do comportamento e personalidade, sinais neurológicos focais e, por vezes, sinais de hipertensão intracraniana; trata-se da forma mais grave e frequente (~ 50% dos casos);
- Pneumonia, geralmente assintomática ou ligeira, com sintomas inespecíficos como tosse, toracalgia, dificuldade respiratória, perda ponderal e fadiga (~ 30% dos casos);
- Doença grave disseminada (em geral nos doentes com SIDA), assumindo um quadro clínico símile septicémia com disfunção multiorgânica e prognóstico reservado;
- Infecção cutânea (lesões nodulares, podendo ulcerar, pústulas ou celulite) (Figura 3);
- Infecção esquelética com compromisso frequente das vértebras;
- Infecção ocular (coriorretinite);
- Linfadenopatia generalizada.
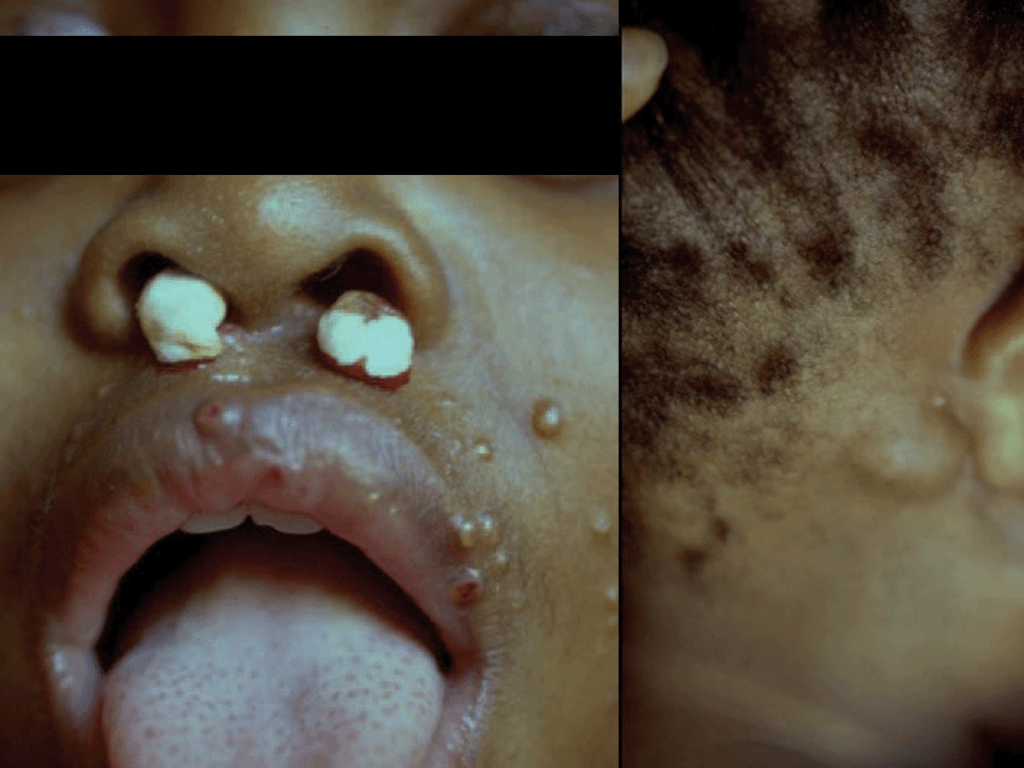
FIGURA 3. Lesões cutâneas nodulares de Criptococose [regiões nasal (ulceradas), labial, geniana, auricular e retroauricular]. (NIHDE)
Diagnóstico
O diagnóstico específico depende da demonstração do fungo (observação directa com microscópio, ou após cultura) ou do seu antigénio capsular no LCR, sangue, ou outros locais atingidos pela infecção; salienta-se, no entanto, que o C. neoformans pode estar presente na expectoração na ausência de doença.
O exame do LCR poderá não revelar alterações, sobretudo nos doentes com SIDA. Nos estudos pediátricos verificou-se que a celularidade é normal em 50% dos casos, podendo variar entre 10-300/mm3 nos restantes casos; existe sempre predomínio de linfócitos.
Habitualmente a glicorráquia e proteinorráquia são também normais. No LCR a formação de um halo com coloração de tinta da china tem uma sensibilidade de 50% a 80%.
No mercado existem kits que permitem a detecção do antigénio capsular no soro ou LCR, com uma sensibilidade e especificidade de 95% no LCR. Falsos negativos surgem quando as concentrações do antigénio são muito baixas ou muito elevadas (Prozona), a estirpe não é capsulada ou o doente é ligeiramente pouco imunodeprimido.
A radiografia do tórax pode evidenciar opacidades nodulares e/ou linfadenopatia hilar.
Na meningoencefalite criptocócica as alterações imagiológicas são inicialmente parcas. Através da RM, pode verificar-se captação meníngea em 15% dos casos. Ocasionalmente pode surgir hidrocefalia, edema cerebral ou massas granulomatosas denominadas criptococomas.
Diagnóstico diferencial
Perante quadro de infecção sistémica acompanhada de meningite, o diagnóstico diferencial inclui investigar, entre diversas causas de meningite, em particular, a tuberculose. As lesões cutâneas impõem que o mesmo se faça com molluscum contagiosum e histoplasmose, designadamente nos doentes com SIDA.
Tratamento e prognóstico
- Nos doentes com doença do sistema nervoso central ou outra forma grave associa-se a anfotericina B convencional (1 mg/kg/dia) ou lipossómica (3 a 6 mg/kg/dia) com a 5-flucitosina (100 mg/kg/dia, para níveis séricos entre 30 a 80 μg/mL) durante duas semanas (terapêutica de indução), seguida de fluconazol 12 mg/kg/dia durante 8 semanas (máximo 800 mg; terapêutica de manutenção); deve ser realizada punção lombar após duas semanas para documentar a esterilização. Nos doentes com cultura positiva às 2 semanas a terapêutica deverá ser prolongada.
- Nos casos de disfunção imunitária (SIDA e outras) em que são frequentes as recaídas após paragem do tratamento deverá manter-se a terapêutica supressiva com fluconazol (6 a 10 mg/kg/dia).
- Nos casos de doença pulmonar sintomática: fluconazol 10 a 12 mg/kg/dia, sendo a duração do tratamento guiada pela evolução clínica, serológica e radiográfica. As novas gerações de azóis (voriconazol e posaconazol) são também efectivas contra a infecção por Cryptococcus; tal não acontece, no entanto, com as equinocandinas (micafungina e caspofungina).
Com a terapêutica antiretrovírica associada nos casos de SIDA o prognóstico melhorou. Uma das complicações da meningite por criptococose é a hidrocefalia obstrutiva.
Prevenção
A administração profiláctica de fluconazol em doentes com infecção por VIH poderá reduzir significativamente o risco de criptococose; contudo, na prática, este tipo de quimioprofilaxia não está indicado, tendo em conta a reduzida prevalência de tal patologia fúngica associada.
2.4 Blastomicose
Importância do problema
A blastomicose é uma doença fúngica rara causada por Blastomyces dermatitidis, fungo dimorfo (micelar quando na natureza, e leveduriforme, com cápsula espessa, quando nos tecidos). Trata-se duma doença endémica, rara em idade pediátrica (abaixo dos 15 anos, corresponde a ~ 2%-10% dos casos notificados em todas as idades). Estão descritos casos em todos os continentes, sendo a maioria dos casos da região central da América do Norte, alguns locais de África e Médio Oriente. O agente, difícil de isolar no solo, aparece sobretudo em cursos de água. A doença adquire-se por inalação de esporos (na espécie humana e em animais), não se transmitindo de pessoa a pessoa. A infecção também pode ser adquirida através de lesão cutânea.
Etiopatogénese
O local primário de infecção é, em geral, o pulmão. Os esporos, atingindo os alvéolos, iniciam germinação, passando a formas leveduriformes. Embora, na maioria das vezes, os esporos sejam fagocitados pelos macrófagos antes de surgir infecção, os que sobrevivem originam pneumonite, podendo seguir-se disseminação hematogénica. Como resposta imune à invasão do agente, neutrófilos e macrófagos migram para os tecidos infectados. O resultado final é uma resposta piogranulomatosa associada a necrose e ulterior fibrose. As lesões cutâneas podem ser secundárias a disseminação hematogénica ou a inoculação directa.
Manifestações clínicas
A infecção por Blastomyces dermatitidis pode ser assintomática, autolimitada, e, por isso, não diagnosticada. Os sinais e sintomas são muito variáveis, entre: pneumonia aguda autolimitada associada a sinais gerais e inespecíficos como tosse, febre, perda ponderal, cefaleias, dor abdominal, sudorese nocturna; e quadro de doença aguda, crónica ou fulminante.
De salientar que a pneumonia aguda por blastomicose poderá não regredir espontaneamente e manifestar-se por vezes como infecção pulmonar subaguda ou crónica, manifestando-se como perda de peso, astenia e tosse, semelhante à tuberculose, sarcoidose e histoplasmose. Por vezes acompanha-se de eritema nodoso. A apresentação como nódulos ou derrame pleural é muito menos frequente.
As manifestações extrapulmonares incluem, entre outras, alterações cutâneas (a manifestação extrapulmonar mais frequente, sob a forma de lesões verrugosas ou ulcerações), lesões osteoarticulares (osteomielite), génito-urinárias e do sistema reticuloendotelial (fígado, baço, gânglios linfáticos, medula).
Diagnóstico
A blastomicose, embora rara na idade pediátrica, deve ser suspeitada em doentes vivendo em áreas endémicas, com lesões granulomatosas e ulceradas da pele ou mucosas, tendendo para a cronicidade. A forma crónica não se distingue da tuberculose, histoplasmose ou coccidioidomicose.
O diagnóstico definitivo faz-se através de exame directo em microscopia e de isolamento por cultura do fungo a partir das lesões (tecidos e fluidos corporais infectados).
É possível a detecção de antigénio específico na urina (método ELISA, por ex.) mas existem reacções cruzadas com outros fungos; um resultado negativo não exclui infecção. Os métodos serológicos têm uma sensibilidade baixa e não devem ser usados.
O exame radiográfico do tórax pode evidenciar sinais de consolidação, infiltrados intersticiais e alveolares, geralmente sem cavitação.
Tratamento
O tratamento da blastomicose depende da gravidade da infecção, envolvimento do SNC e a integridade do sistema imune. O tratamento é recomendado para todos os doentes pelo risco de disseminação.
A anfotericina B convencional (0,7-1 mg/kg/dia) ou a anfotericina B lipossómica (3-5 mg/kg/dia, preferida se houver envolvimento do SNC) são recomendadas na terapêutica inicial da doença grave; nas formas moderadas a ligeiras ou após estabilidade clínica nas formas graves recomenda-se itraconazol oral (10 mg/kg/dia). O tratamento deverá durar 6-12 meses, sendo no mínimo de 12 meses nas infecções do SNC e osteomielite.
2.5 Coccidioidomicose
Importância do problema
A coccidioidomicose, também designada por febre de São Joaquim (nome derivado do vale de São Joaquim na Califórnia, onde a doença tem alta prevalência), é uma doença causada por um fungo dimórfico (Coccidioides immitis), existindo no solo sob a forma micelar/filamentosa, e nos tecidos sob a forma esporular. Uma segunda espécie (C. posadasii) com idêntica patogenicidade foi isolada em áreas fora da Califórnia.
Endémica em certas regiões da América do Norte, México, América Central e América do Sul incluindo Brasil, pode afectar hospedeiros com e sem imunodeficiência. A doença confere imunidade permanente (resposta TH1 ou T helper 1).
Etiopatogénese
A doença adquire-se através das conídias na fase micelar saprofítica (altamente infecciosas) que podem ser inaladas com a poeira do solo ou penetrar na pele em que se verifica solução de continuidade. Após inalação pode surgir doença cerca de 3 dias depois, coincidindo com a evolução para a forma esporular (esférula endosporo). A reacção tecidual é inflamatória, com influxo de neutrófilos e formação de granuloma. Não existe transmissão pessoa a pessoa (exceptuando a eventualidade de através de órgãos transplantados), mas foi descrita transmissão vertical mãe-feto.
Manifestações clínicas
Na maioria dos casos a sintomatologia é semelhante à de síndroma gripal (cefaleias, febre, artralgias e mialgias), por vezes associada a pneumonia, com recuperação espontânea. Como sequelas pulmonares poderão aparecer ocasional e paulatinamente nódulos ou cavidades, sobretudo em doentes com diabetes; tal achado pode verificar-se após radiografia do tórax, o que demonstra que tal evolução é assintomática ou oligossintomática.
A infecção aguda pode estar associada a exantema, eritema multiforme, assim como eritema nodoso. Verifica-se disseminação extrapulmonar (osteoarticular, do SNC, renal e cutâneo) em 0,5% dos casos, associada a imunodeficiência. Os locais mais frequentes de disseminação são a pele, o sistema osteoarticular e o sistema nervoso central.
Diagnóstico
O diagnóstico pode ser confirmado por:
- Exame microscópico directo e por cultura da expectoração, pus ou sangue;
- Provas serológicas (fixação do complemento, EIA) para doseamento de IgG e IgM; em 50% a 90% das infecções primárias a IgM é positiva entre a primeira e a terceira semana de doença; títulos de IgG > 1/16 estão em geral associados a infecções mais graves;
- Detecção do antigénio na urina, soro ou lavado broncoalveolar, evidenciando uma sensibilidade mais elevada na doença grave (podem ocorrer reações cruzadas);
- Exame radiográfico do tórax poderá evidenciar sinais de infiltrados com adenopatia hilar, de derrame ou de cavitações (nas formas complicadas).
O diagnóstico diferencial faz-se essencialmente com a tuberculose, tendo em consideração o quadro febril, os achados radiológicos torácicos e o eritema nodoso.
Tratamento e prognóstico
A coccidioidomicose primária raramente exige tratamento que não seja sintomático.
O tratamento específico (que engloba vários esquemas) deve ficar reservado para as formas graves associadas a títulos de anticorpos fixadores do complemento IgG > 1/16 (pós-primárias, primárias graves, disseminadas, ou de evolução subaguda ou crónica).
Nestes casos recomenda-se fluconazol (10 mg/kg/dia), ou itraconazol (nível > 1 μg/ml) PO 2,5-5 mg/kg/ dia, 3 a 6 meses.
A anfotericina B lipossómica está indicada nas formas disseminadas, em doses até 3-5 mg/kg/dia. Se houver compromisso do SNC pode ser dada também por via intratecal (0,1-0,5 mg/kg/dia), designadamente se se tratar de C. immitis. O tratamento deve durar até resolução da sintomatologia, em regra, e no mínimo, ~ 6 meses.
O prognóstico da coccidioidomicose é excelente, o da pós-primária é bom, e o da forma disseminada é muito reservado a mau, sobretudo se houver infecção meníngea.
Prevenção
A prevenção diz respeito à evicção da exposição a conídias. Uma vez que as vacinas holocelulares mortas são ineficazes, presentemente está em fase de investigação uma vacina subcelular.
2.6 Histoplasmose
Importância do problema
A histoplasmose é uma infecção pelo fungo dimórfico Histoplasma capsulatum cujas formas infectantes são as macro e microconídeas (esporos) das formas micelares; as mesmas encontram-se no solo rico em nitratos, com dejectos de aves e morcegos, e em zonas de pó com prédios e madeiras em ruínas. Os esporos podem ser transportados nas asas das aves.
Esta infecção tem distribuição mundial (estimando-se cerca de 200 000-500 000 casos anuais), sendo endémica nas regiões oriental e central dos EUA e América Latina. Na Europa e Ásia têm sido descritos casos esporádicos. Pode afectar crianças com e sem imunodeficiência. Não se transmite de pessoa a pessoa.
Etiopatogénese
A infecção surge pela inalação das microconídeas. Nos pulmões, os esporos germinam, evoluindo para formas leveduriformes que, provocando influxo de neutrófilos, linfócitos e macrófagos, levam à formação de granulomas. As formas leveduriformes podem sobreviver nos macrófagos e sistema reticuloendotelial durante anos.
As anomalias primárias ou adquiridas da função de imunidade celular, assim como a imaturidade relativa da imunidade celular na primeira infância, são factores de risco quanto a disseminação do microrganismo.
Verificando-se disfunção dos linfócitos T, o foco infeccioso inicial pode expandir-se e disseminar-se. A probabilidade de infecção é directamente proporcional à carga de inóculo, sendo que aquela poderá ser assintomática e autolimitada em 10%-50% dos casos.
Manifestações clínicas
Na maioria dos casos as infecções são subclínicas ou autolimitadas, não requerendo confirmação laboratorial.
Quando sintomática, a infecção pode ser pulmonar, extrapulmonar ou disseminada; e ainda, aguda ou crónica. Na maioria, apresenta-se sob a forma pulmonar aguda, com sintomas ligeiros (febre, tosse, mal-estar geral, adenopatia hilar e escassos infiltrados pulmonares). Nalguns casos pode surgir também envolvimento do mediastino, artrite, pericardite ou eritema nodoso.
Nas áreas endémicas pode ser observada uma forma cutâneo-mucosa com formação de granulomas. Nas referidas áreas endémicas, em crianças com aparente bom estado geral, uma radiografia torácica eventualmente realizada, evidenciando sinais compatíveis com granulomas típicos, poderá determinar a realização de exames laboratoriais.
A histoplasmose disseminada é uma doença progressiva mais rara, cujo foco inicial tem como ponto de partida a infecção primária do pulmão. Surgindo quase exclusivamente em crianças com imunodeficiência, pode seguir-se à infecção aguda, ou manifestar-se anos mais tarde, com febre prolongada (semanas ou meses), hepatosplenomegália progressiva, choque e disfunção multiorgânica com insuficiência hepática, renal, aplasia medular, compromisso do SNC e CIVD (quadro septicémico).
Diagnóstico
Dum modo geral, a realização de exames laboratoriais poderá ser necessária em doentes sintomáticos com quadro clínico podendo sugerir outros agentes patogénicos tais como Mycobacterium tuberculosis, Blastomyces dermatiditis, ou outros, susceptíveis de provocar inflamação granulomatosa.
O diagnóstico implica um elevado índice de suspeita numa criança que viva (ou tenha estado) em zona endémica e a eventual realização dum conjunto de exames complementares a saber:
- Exame cultural para isolamento do agente (sangue, LCR, urina, lavado broncoalveolar, biópsias de tecidos infectados);
- Detecção de antigénio por método ELISA (soro, urina, lavado broncoalveolar);
- Detecção de anticorpos/fixação do complemento (sendo possível surgir reacções cruzadas); títulos > 1/32 ou aumento superior a 4 vezes o título cerca de 4-6 semanas após exposição ao fungo sugerem o diagnóstico.
O diagnóstico diferencial, tendo em conta a doença pulmonar e o padrão radiográfico do tórax, faz-se fundamentalmente com a tuberculose miliar.
Tratamento
Verificando-se a infecção pulmonar primária nos doentes sem imunodeficiência, não está indicado tratamento específico em tais circunstâncias. Como se pode depreender, estão indicadas medidas habituais de suporte para as infecções das vias respiratórias inferiores como oxigenoterapia, fluidoterapia, etc..
Não se verificando melhoria clínica do quadro de infecção pulmonar ao final de 1 mês, está indicado o tratamento com itraconazol 5 mg/kg/dia 6 a 12 semanas (níveis séricos > 1 μg/ml). Nos casos pulmonares graves sugere-se anfotericina B lipossómica (3 mg/kg/dia) IV durante 1-2 semanas, seguida de itraconazol PO (2-5 mg/kg/dia) durante 12 semanas. Nas crianças com imunodeficiência, a terapêutica poderá ser mais prolongada, até 1 ano.
Nas crianças com imunodeficiência, designadamente com infecção por VIH vivendo em zonas endémicas, poderá ser considerada a quimioprofilaxia com itraconazol (2-5 mg/kg cada 12 ou 24 horas).
A corticoterapia, num período curto e sempre concomitantemente com a terapêutica anti-fúngica, deverá ser reservada para as situações com alteração ventilatória secundária a adenopatias importantes, levando a obstrução da via respiratória.
2.7 Pneumocistose
Importância do problema
A infecção por Pneumocystis jiroveci (anteriormente designado P. carinii) origina um quadro de pneumonia intersticial (sigla habitual do inglês – PCP, significando Pneumocystis pneumonia) no contexto de determinados factores predisponentes (imunodeficiência), na maioria dos casos em crianças antes dos 4 anos. Nos doentes imunocompetentes a infecção é geralmente subclínica e não diagnosticada.
Mesmo nas formas mais graves de infecção, com raras excepções, a doença localiza-se no pulmão. Actualmente, este agente é classificado como fungo e não como protozoário (com base na análise da sequenciação do DNA), apesar de possuir diversas semelhanças morfológicas e biológicas com os protozoários.
Uma vez que o microrganismo pode também infectar outras espécies animais, designadamente mamíferos, alguns autores continuam a utilizar a nomenclatura P. carinii seguida da sigla f.sp (forma specialis) para designar especificamente a infecção em determinados hospedeiros; por ex. P. carinii f.sp.ratti, P. carinii f.sp muris, P. carinii f.sp. hominis, etc..
Aspectos epidemiológicos e etiopatogénese
O microrganismo está distribuído por todo o mundo. De acordo com estudos serológicos, a maior parte das pessoas é infectada antes dos 2 anos de idade; as infecções em crianças imunocompetentes são geralmente assintomáticas, demonstrando-se a presença de anticorpos em cerca de 75% dos casos.
Os factores predisponentes de pneumonia são: imunodeficiência congénita ou adquirida (designadamente infecção por VIH), desnutrição, doenças do foro oncológico, doentes submetidos a transplante de órgãos, corticoterapia, terapêutica imunossupressora, sobretudo com anti-TNF, etc.. Segundo alguns estudos, o microrganismo foi isolado nos pulmões de lactentes com síndroma de morte súbita, sem se ter concluído sobre a possível relação causa-efeito.
O habitat natural e o modo de transmissão ao homem são desconhecidos. A transmissão entre animais faz-se por via inalatória, sendo provável que a transmissão inter-humana se faça da mesma maneira; a transmissão animal-homem é pouco provável pelo facto de determinadas espécies do agente infectarem determinadas espécies de hospedeiros (ver atrás forma specialis).
Nos espaços alveolares encontram-se 2 formas de P. jiroveci (cuja terminologia deriva da similitude com a morfologia dos protozoários): quistos com 5-8 um de diâmetro contendo esporozoítos intraquísticos; e trofozoítos extraquísticos. O microrganismo, atingido o alvéolo, adere aos pneumatócitos de tipo I com o auxílio de proteínas adesivas como a fibronectina.
A capacidade de o agente provocar lesão anátomo-patológica pulmonar depende fundamentalmente da normalidade dos mecanismos de imunidade celular. Com efeito, em estudos realizados em doentes com SIDA, verificou-se aumento da incidência de pneumonia relacionada com a diminuição do número de linfócitos T CD4+ (sobretudo no grupo etário 3-6 meses).
Admite-se que os referidos linfócitos tenham papel importante na depuração dos microrganismos interagindo com fagócitos, complemento, e activação dos macrófagos; em caso de disfunção deste processo, produz-se lesão inflamatória conduzindo a destruição do surfactante, entre outros efeitos.
As consequências anátomo-patológicas são a génese de 2 quadros morfológicos: pneumonite intersticial de plasmócitos (sobretudo em lactentes com desnutrição, em que predominam plasmócitos no processo inflamatório, e se verifica infiltração com espessamento dos septos alveolares); e pneumonite alveolar descamativa difusa (sobretudo em crianças e adultos com imunodeficiência, em que há exsudado alveolar sem compromisso dos septos alveolares, e sem plasmócitos).
De salientar que a quimioprofilaxia associada a terapêutica anti-retrovírica activa na actual chamada “era HAART “(highly active antiretroviral therapy), contribuíram para reduzir significativamente o nº de casos/100 doentes-ano: de 5,8 (anteriormente) para 0,3.
Manifestações clínicas
As manifestações clínicas, em geral de início insidioso, incluem febre, dificuldade respiratória de grau variável, tosse não produtiva e sinais de hipóxia. Em geral, os sinais auscultatórios são discretos ou ausentes. A intensidade dos sinais e sintomas varia, podendo ser aguda ou fulminante, dependendo da imunossupressão do doente. (Figura 4)
Exames complementares e diagnóstico
A radiografia do tórax revela infiltrado intersticial difuso ou alveolar e, mais raramente, lesões lobares, miliares ou nodulares; contudo, pode não surgir qualquer alteração radiológica nas fases iniciais da doença. (Figura 5)
Para o diagnóstico etiológico, torna-se fundamental identificar o agente (por diversas técnicas como imunofluorescência, por técnica altamente específica e sensível empregando anticorpo monoclonal conjugado com fluorescência, ou PCR, com boa sensibilidade, mas especificidade inferior a 85%-90%) no lavado broncoalveolar, em expectoração induzida (crianças mais velhas), ou por métodos invasivos (por ex. biópsia pulmonar).
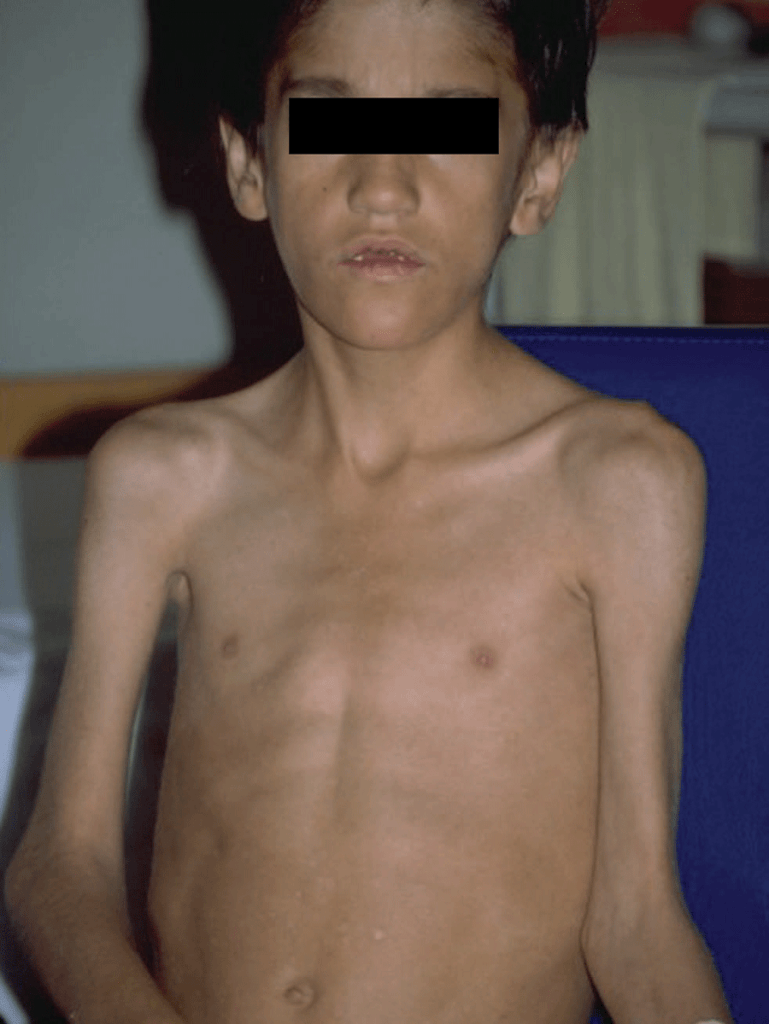
FIGURA 4. Criança com SIDA. Aspecto geral emagrecido associado a pneumonia por P. jiroveci. (NIHDE)
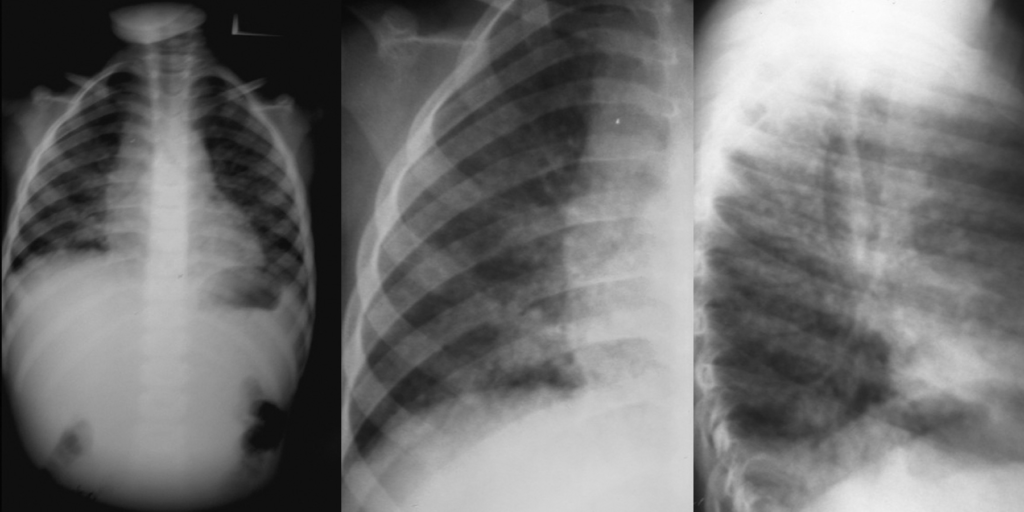
FIGURA 5. Padrão radiológico de pneumonia por P. jiroveci no contexto de SIDA/VIH. Opacidades nodulares confluentes ocupando o ⅓ inferior do campo pulmonar direito (incidências póstero-anterior e perfil direito). (NIHDE)
Tratamento
- TMP/SMX – ( TMP 15-20 mg/kg/dia; SMX 75-100 mg/kg/dia, dividida em 3 a 4 doses) PO ou IV, durante 14-21 dias – o tratamento de eleição; como alternativa, nos doentes que não toleram o TMP/SMX, ou em que se verifica falência terapêutica, poderá optar-se por:
- Pentamidina na dose de 4 mg/kg/dia IV, durante 14-21 dias (pode ser alterada para atovaquona ao fim de 7 a 10 dias);
- Atovaquona PO, na doença ligeira a moderada, 30-40 mg/kg/dia (se 1-3 meses de idade ou > 24 meses); 45 mg/kg/dia (se 4-24 meses de idade) durante 14-21 dias.
- Corticóides – foram comprovados benefícios do seu emprego em adolescentes e adultos infectados por VIH e com pneumonia por PCP moderada a grave. Nas crianças, contribuem para reduzir a gravidade da insuficiência respiratória aguda, a necessidade de ventilação e a mortalidade. As doses recomendadas de prednisolona oral (em idades > 13 anos) são: 80 mg/dia divididas em duas doses, nos primeiros 5 dias, 40 mg/dia de D6-D10, e 20 mg/dia D11-D21. Nas crianças mais novas (< 13 anos): 1 mg/kg/dose de 12/12 horas durante 5 dias; 0,5 mg/kg/dose de 12/12 horas de D6-D10 e 0,5 mg/Kg/dia, dose única de D11-D21.
- Tratando-se duma pneumonia, tal obriga a medidas de suporte geral abordadas na Parte sobre Pneumologia.
Quimioprofilaxia
A verificação de determinados factores de risco estabelecem a indicação de quimioprofilaxia: doentes imunodeprimidos com um episódio prévio de pneumonia por Pneumocystis jiroveci, crianças com imunodeficiência celular grave, receptores de transplante de órgãos, doenças linfoproliferativas ou outro tipo de neoplasias que requerem quimioterapia intensa, crianças com infecção VIH suspeita ou confirmada.
Nestes últimos, deve ser efectuada profilaxia desde as 4 a 6 semanas até um ano de idade, até exclusão de infecção VIH (por ex. lactentes de mães com infecção por VIH).
Após um ano de idade, a necessidade de quimioprofilaxia é orientada pela percentagem e número de linfócitos T CD4+. O fármaco de primeira linha é o TMP-SMX (respectivamente 5 mg/kg – 25 mg/kg/dia), dividido em duas tomas diárias, 3 dias por semana, consecutivos). A pentamidina aerossolizada, a atovaquona e dapsona são alternativas de segunda linha se o TMP-SMX não for tolerado.
Prognóstico
O prognóstico da pneumonia por Pneumocystis jiroveci depende da imunodeficiência subjacente, comporta mortalidade que poderá oscilar entre 25% e 40% nos doentes com neoplasias e 5% a 10% nos doentes com SIDA. Sem tratamento é, em geral, fatal.
2.8 Outras infecções fúngicas sistémicas
Citam-se, por fim, e de modo sucinto, dois tipos de infecções fúngicas sistémicas, mais raras, as quais fazem parte duma lista mais vasta:
- Paracoccidioidomicose (Blastomicose sul-americana) causada por Paracoccidioides brasiliensis;
- Micetoma eumicótico, tendo como agentes mais frequentes Madurella mycetomatis (70%) e Pseudallerscheria boydii e Leptosphaeriae senegalensis (10%), entre outros.
BIBLIOGRAFIA
American Academy of Pediatrics/AAP. Red Book 2015. Report of the Committee on Infectious Diseases. Elk Grove Village, IL: AAP, 2015
Antachapoulos C, Walsh TJ. New agents for invasive mycoses in children. Curr Opin Pediatr 2005;17:78-87
Bennett JF, Dolin R, Blaser MJ (eds). Mandell, Douglas and Bennett’s Principles and Practice of Infectious Diseases. Philadelphia: Elsevier, 2015
Bergelson JM, Shah SS, Zaoutis TE. Pediatric Infectious Diseases. The Requisites in Pediatrics. Philadelphia: Mosby Elsevier, 2008;297-309
Blyth C C. Palasanthiran P, O´Brian TA. Antifungal therapy in children with invasive fungal infections: a systematic review. Pediatrics 2007;119:772-784
Esteves JA, Baptista AP, Rodrigo FG, Gomes MA. Dermatologia. Lisboa: Fundação Calouste Gulbenkian, 1992
Feigin RD, Cherry JD, Demmler GL, et al (eds). Textbook of Pediatric Infectious Diseases. Philadelphia: Saunders, 2014
Ferreira WFC, Sousa JC, Lima N (eds). Microbiologia. Lisboa: Lidel, 2010
Garcia JJ, Cruz O, Mintegi S, Moreno JM (eds). M Cruz Manual de Pediatria. Madrid: Ergon, 2020
Goldman L, Schafer AI (eds). Goldman – Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Guerra-Rodrigo F, Marques Gomes M, Mayer-da-Silva A, Filipe P I. Dermatologia. Lisboa: Fundação Calouste Gulbenkian, 2010
Helfaer MA, Nichols DG. Roger´s Handbook of Pediatric Intensive Care. Philadelphia: Wolters Kluwer / Lippincott Williams & Wilkins, 2009
Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, et al. A higher-level phylogenetic classification of the Fungi. Mycol Res 2007;111:509 -547
Hope W, Castagnola E, Groll A. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: prevention and management of invasive infections in neonates and children caused by Candida spp. Clin Microbiol Infect 2012;18 (Suppl. 7):38-52
Hernandez T, Machado S, et al. Tinhas do couro cabeludo na idade pediátrica. Nascer e Crescer 2004;13:23-26
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: McGraw Hill Education, 2018
Long SS, Prober CG, Fischer M (eds). Principles and Practice of Pediatric Infectious Diseases. Philadelphia: Elsevier, 2018
MacDonald MG, Seshia MMK (eds). Avery’s Neonatology: Pathophysiology and Management of the Newborn. Philadelphia, PA: Lippincott Williams & Wilkins, 2015
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Pickering LK, Prober CG (eds). Principles and Practice of Pediatric Infectious Diseases. Philadelphia: Churchill – Livingstone – Elsevier, 2012
Sethi A, Antaya R. Systemic antifungal therapy for cutaneous infections in children. Pediatr Infect Dis J 2006;25:643-644
Steinbach WJ, Walsh T J. Mycoses in pediatric patients. Infect Dis Clin North Am 2006;20:663-678