1. CUTIS LAXA
Definição
O termo cutis laxa engloba um grupo heterogéneo de doenças raras, originadas por défice metabólico da síntese das fibras elásticas do tecido conjuntivo, ou por sua alteração degenerativa proteolítica, facto que justifica a designação de elastólise ou de elastorrexis generalizada. Tem sido também designada dermatocalázio.
Existem formas congénitas determinadas geneticamente (autossómicas dominantes, autossómicas recessivas ou ligadas ao cromossoma X), de aparecimento mais precoce, e formas adquiridas.
Etiopatogénese e manifestações clínicas
Descrevem-se as seguintes formas congénitas:
- A mais frequente, mais grave e de início precoce transmite-se de modo autossómico recessivo, compreendendo: – o tipo relacionado com mutação dos genes da fibulina 4 e fibulina 5 (FBLN4 e FBLN5); e – o tipo relacionado com mutação do gene ATP6V0A2;
- A forma autossómica dominante, relacionada com mutação do gene da elastina (ELN em 14q32.1, 7q11.2);
- A forma autossómica dominante ou recessiva resultante de mutação do gene da fibulina 5 (FBLN5 ou DANCE, sigla de Developmental Arteries and Neural Crest EGF like em 14q32.1);
- A forma ligada ao cromossoma X (adenosina trifosfatase transportadora do cobre (ATP7A em Xq12-q13).
Quanto às formas adquiridas, há a referir que podem manifestar-se na infância ou no adulto, sendo em geral, desencadeadas por factores precipitantes gerais ou locais (processos inflamatórios da pele, tais como lúpus eritematoso, amiloidose, eritema multiforme, urticária, angioedema, a reacções de hipersensibilidade à penicilina e, em RN, a ingestão materna de penicilina durante a gravidez).
A manifestação fundamental é o desenvolvimento de grandes pregas cutâneas laxas, sendo que os quadros clínicos variam como resultado do compromisso das fibras elásticas e também dos órgãos atingidos. (Figura 1)
Como sinais mais típicos destacam-se, pois, grandes pregas dérmicas laxas (resultantes de perda de elasticidade da pele), aspecto de envelhecimento prematuro, voz de baixa tonalidade e disfonia por alteração da elasticidade das cordas vocais.
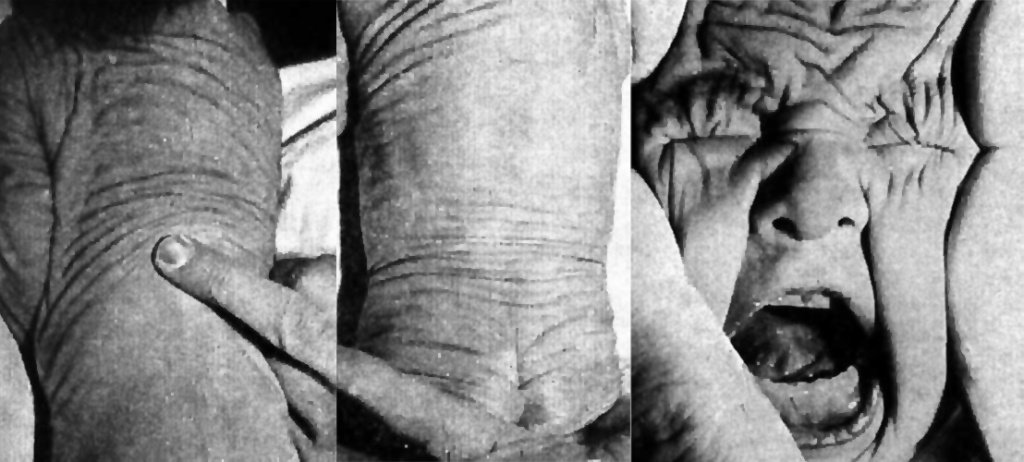
FIGURA 1. Cutis laxa em lactente: pregueamento da pele mantido ao cessar o estiramento
Os sinais viscerais incluem hérnias (Figura 2), divertículos, prolapso vaginal ou rectal, enfisema pulmonar, aneurismas aórticos e/ou estenose da artéria pulmonar. Outros achados englobam: antecedentes de restrição de crescimento intrauterino, baixa estatura, alterações esqueléticas e cárie dentária grave. Os doentes falecem habitualmente na idade adulta jovem.
A cutis laxa pode estar associada a síndromas diversas, tais como síndroma de Barsy, de Lenz Majewski, nanismo hiperostótico, etc..
As formas adquiridas limitam-se geralmente ao tegumento cutâneo.
Diagnóstico diferencial
De acordo com o exame histológico, a presença de pele flácida corresponde à verificação microscópica de diminuição, fragmentação e distensão das fibras elásticas dérmicas e, ocasionalmente, viscerais.
O diagnóstico diferencial, por vezes difícil, faz-se com o pseudoxantoma elástico e as síndromas de Ehlers-Danlos.
Tratamento
Não existe tratamento específico; em casos seleccionados pode estar indicada a cirurgia plástica com resultados variáveis, existindo a probabilidade de recidiva local.
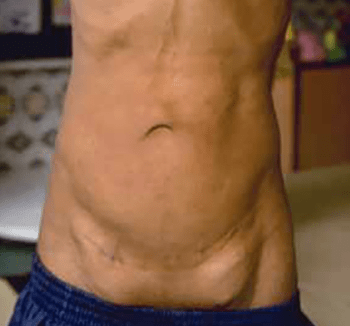
FIGURA 2. Aspecto de fenótipo da cutis laxa congénita em adolescente; são notórias hérnias inguniais. (NIHDE)
2. PSEUDOXANTOMA ELÁSTICO
Definição e etiopatogénese
O pseudoxantoma elástico (ou síndroma de Gronblad-Strandberg), afecção multissistémica hereditária heterogénea rara, caracterizada por alteração da elastina e calcificação das estruturas elásticas dérmicas e vasculares. Com uma frequência de cerca de 1/100.000 nados-vivos, está associada a mutação no gene ABCC6, localizado no braço curto do cromossoma 16 (16p13.1). Transmite-se, na sua maioria, de forma autossómica recessiva.
Admite-se que da mutação genética resulte produção de substâncias (provavelmente glicosaminoglicanos) com afinidade particular para as fibras elásticas. A modificação estrutural destas fibras, com depósito secundário de sais de cálcio, conduz povavelmente às alterações observadas na derme, nas camadas média e íntima de artérias de médio calibre, e na membrana de Bruch do olho.
Manifestações clínicas
As manifestações iniciam-se em geral na adolescência: aparecimento de lesões cutâneas xantomatosas (pápulas pequenas, levemente salientes, da cor da pele ou amareladas), assintomáticas, situadas nas superfícies laterais do pescoço e/ou axilas, bilaterais, por vezes com padrão linear ou reticulado, e nas áreas de flexão. Em cerca de 80% dos casos, observam-se na mucosa oral, superfície interna dos lábios e abóbada palatina.
De salientar ainda, ao nível do olho: estrias angióides detectáveis por fundoscopia, ruptura da membrana elástica de Bruch, hemorragias, degenerescência macular e neovascularização retiniana.
Pode verificar-se também compromisso valvular e arteriopatia oclusiva dos grandes vasos com hipertensão secundária.
A esperança de vida está diminuída por complicações vasculares, sendo que as alterações do globo ocular poderão conduzir à amaurose.
Diagnóstico
O diagnóstico é confirmado pela presença das denominadas estrias angióides no exame fundoscópico; embora tal padrão esteja presente em cerca de 85% dos casos, não é patognomónico da afecção (podem detectar-se também na síndroma de Ehlers-Danlos e na doença de células falciformes.
A biópsia dérmica evidenciando sinais de depósitos cálcicos em fibras elásticas fragmentadas, degeneradas e distorcidas pode corroborar o diagnóstico.
O diagnóstico diferencial estabelece-se fundamentalmente com o líquen escleroatrófico e a esclerodermia.
Tratamento
Não existe tratamento curativo, podendo, no entanto, ser combatidos os factores de risco cardiovascular, assim como proceder-se ao tratamento sintomático das complicações.
Nas situações de compromisso valvular mitral significativo poderá estar indicada intervenção de cirurgia cardiovascular.
Nas formas de manifestações oculares poderá estar indicada a terapêutica com laser; com o objectivo de tentar reduzir a taxa de hemorragias retinianas têm sido utilizados suplementos nutricionais (A, C, E, ácidos gordos essenciais como por ex. ómega 3, etc.) e de oligoelementos (Zn, Se) cuja eficácia é duvidosa.
3. SÍNDROMA DE WILLIAMS
Definição e etiopatogénese
Trata-se duma rara afecção caracterizada essencialmente por problemas cardíacos e do tecido conectivo (por alteração da síntese da elastina), e devida a microdeleção de genes contíguos (gene da elastina e LIMK1) do braço longo do cromossoma 7q11.23. É transmitida hereditariamente segundo o modo AD, com uma frequência de 1/ 20.000 indivíduos.
Manifestações clínicas
Como manifestações fundamentais são descritas: fácies dismórfica típica de “duende” (crânio com depressão bitemporal, hipoplasia facial média, hipertelorismo, nariz arredondado, má oclusão dentária, dentes com hipoplasia do esmalte, baixa estatura, défice ponderal, e défice cognitivo moderado e comportamento peculiar (designadamente grande sociabilidade e capacidade auditiva-musical muito notória).
Outras manifestações incluem: arteriopatia (estenose pulmonar, estenose aórtica supravalvular), hiperlaxidão, hipotiroidismo, contracturas articulares, hipercalcémia, hipercalciúria, nefrocalcinose, hipotonia, etc..
Para o diagnóstico são utilizadas técnicas de hibridação.
Tratamento
Não existe tratamento específico; as medidas a aplicar incluem tratamento sintomático e reabilitação com o objectivo de integração social.
BIBLIOGRAFIA
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Goodman RM, Gorlin RJ. The Malformed Infant and Child. An Illustrated Guide. Oxford: Oxford University Press, 1983
Guerra-Rodrigo F, Marques-Gomes M, Mayer-da-Silva A, Filipe P I. Dermatologia. Lisboa: Fundação Calouste Gulbenkian, 2010
Habif TP (ed). Clinical Dermatology. Philadelphia: Mosby, 2004
Jones KL, Smith DW. The Williams elfin facies syndrome: a new perspective. J Pediatr 1975; 86: 718-720
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
McKusick VA, Amberger JS, Francomano CA. Progress in medical genetics: map-based gene discovery and the molecular pathology of skeletal dysplasias. Am J Med Genet 1996; 63: 98-105
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Myllyharju J, Kivirikko KI. Collagens and collagen-related diseases. Ann Med 2001; 33: 7-21
Ohtani T, Furukawa F. Pseudoxanthoma elasticum. J Dermatol 2002; 29: 615-620
Royce PM, Steinmann B (eds). Connective Tissue and its Disorders: Molecular, Genetic, and Medical Aspects. New York: Wiley-Liss, 2002