Definição e importância do problema
As principais anomalias que definem a tetralogia de Fallot são: obstáculo da câmara de saída do ventrículo direito (por desvio anterior do septo infundibular) e comunicação interventricular (grande, localizada na porção perimembranosa subaórtica do septo).
Classicamente descrevem-se mais duas alterações – hipertrofia do ventrículo direito (secundária ao obstáculo direito) e “cavalgamento aorto-septal” de grau variável (relacionado com o grau de desvio anterior do septo infundibular).
Descrita pela primeira vez em 1888 por Étienne-Louis Arthur Fallot, a tetralogia de Fallot é a cardiopatia congénita cianótica mais frequente após o primeiro ano de vida.
Esta cardiopatia tem uma prevalência de aproximadamente 4-5 em cada 10.000 nados-vivos, correspondendo a cerca de 7-10% de todas as cardiopatias congénitas, sem predomínio de género.
Anatomia
A anomalia embriológica exacta responsável pela tetralogia de Fallot é ainda desconhecida, admitindo-se, contudo, que o achado fisiopatológico patognomónico desta doença seja o desvio anterior e cefálico do septo infundibular. Deste desvio resulta obstáculo da câmara de saída do ventrículo direito e uma comunicação interventricular (CIV) grande, mal-alinhada, localizada à região perimembranosa do septo interventricular, com dextroposição e cavalgamento da raiz da aorta sobre a CIV. A hipertrofia ventricular direita associada a esta doença é secundária à CIV grande e ao obstáculo da câmara de saída do ventrículo direito, resultando em pressões ventriculares direitas sistémicas.
O obstáculo da câmara de saída do ventrículo direito verifica-se habitualmente a múltiplos níveis:
- infundibular, secundário a desvio anterior e cefálico do septo infundibular, associando-se a hipertrofia de bandas musculares;
- valvular pulmonar, por hipoplasia do anel valvular pulmonar e por malformação da válvula pulmonar propriamente dita, frequentemente, bicúspide e estenótica;
- supravalvular pulmonar, a nível do tronco e/ou ramos da artéria pulmonar. O predomínio é infundibular em 45% dos casos, valvular em 10%, infundibular e valvular em 30%.
Podem coexistir outras anomalias anatómicas cardíacas e extracardíacas, que ocorrem em até 40% dos doentes com tetralogia de Fallot. Em 25% dos casos o arco aórtico é direito, e em 9% há alterações das artérias coronárias. O tronco e os ramos da artéria pulmonar são, na maioria dos casos, hipoplásicos. Nalguns casos um dos ramos, habitualmente o esquerdo, não tem continuidade com o tronco pulmonar, sendo irrigado pelo canal arterial e/ou por colaterais aorto-pulmonares.
Comunicações interauriculares, comunicações interventriculares múltiplas, defeito do septo aurículo-ventricular e anomalias de conexão venosa são outras malformações encontradas.
Apesar de estarem descritos diversos genótipos associados a tetralogia de Fallot, na maioria dos casos (~75-80%) a cardiopatia em análise ocorre não associada. De salientar no entanto algumas das situações em que se verifica associação: microdeleção- del 22q11.2 (a mais comum), síndroma de Di George, síndroma de Down (trissomia 21), e síndroma de Alagille (mutação JAG1).
Fisiopatologia
A gravidade do obstáculo da câmara de saída do ventrículo direito, que determina a direcção e magnitude do fluxo sanguíneo através da comunicação interventricular, condiciona o quadro clínico. Quando o obstáculo é ligeiro, predomina o shunt esquerdo-direito, pelo que não há cianose. Nos obstáculos moderados, a resistência à ejecção do ventrículo direito é semelhante à resistência vascular sistémica, pelo que o shunt é bidireccional e não há cianose em repouso. Com o exercício, diminui a resistência vascular sistémica e predomina o shunt direito-esquerdo, com a consequente cianose. Quando o obstáculo é grave, o débito pulmonar está muito reduzido e há shunt direito-esquerdo, com cianose grave.
As crises de hipóxia e a necessidade de o doente adoptar posição de cócoras (squatting) são manifestações importantes da gravidade da doença. As crises de hipóxia ou crises de cianose, mais frequentes entre os seis meses e os dois anos de idade, manifestam-se por episódios paroxísticos de cianose intensa com palidez, hiperpneia, irritabilidade e choro prolongado. Acompanham-se de hipotonia, sonolência ou perda da consciência, e podem provocar convulsões, acidente vascular cerebral ou mesmo morte. Durante as crises, diminuem a intensidade e duração do sopro de expulsão pré-existente (que pode mesmo desaparecer), reaparecendo quando a criança recupera. Em geral, são de curta duração (menos de 15 minutos), ocorrem de manhã ao acordar, e podem ser precipitadas pelo esforço, ambiente quente, ou poderá não haver factor desencadeante. Surgem, tanto nas crianças com cianose persistente, como nas crianças acianóticas.
As referidas crises constituem uma emergência e exigem internamento hospitalar para tratamento imediato. A diminuição da pressão parcial de oxigénio (PaO2) arterial, a acidose e hipercapnia resultantes, além de desencadearem as manifestações neurológicas, facilitam o metabolismo anaeróbio e consequente agravamento da acidémia. Esta, actuando sobre o centro respiratório, agrava a hiperpneia com consequente aumento da resistência vascular pulmonar e perpetuação do círculo vicioso.
A posição de “cócoras” (ou outra) é adoptada por crianças mais velhas com a finalidade de aliviar a cianose, dispneia ou lipotímia induzidas pelo esforço. Admite-se, pois, que a angulação e compressão das artérias femorais provocadas por esta manobra excluem da circulação o sangue insaturado dos membros inferiores e aumenta a resistência vascular periférica. Assim, com a referida postura, promove-se o incremento do fluxo pulmonar e a diminuição da hipoxémia, o que leva a que os doentes se sintam mais confortáveis.
A evolução habitual é a de agravamento do obstáculo de saída do ventrículo direito. Nas crianças não tratadas a cianose aumenta e as crises de hipoxémia tornam-se mais frequentes, mais graves e mais prolongadas, podendo ser fatais. A hipóxia crónica estimula a medula óssea a aumentar a produção de eritrócitos (através da libertação de eritropoietina renal). A policitémia compensatória é útil até que o hematócrito atinja valores perto de 70%. A partir deste valor, o aumento da viscosidade do sangue e a diminuição da capacidade de extracção de O2 aumentam o risco de tromboses arteriais (principalmente pulmonares e cerebrais). Por outro lado, as alterações da coagulação secundárias à hipóxia aumentam o risco de acidente vascular cerebral e de abcesso cerebral, sobretudo se associadas a anemia ferropénica.
Manifestações clínicas
A apresentação clínica do doente com tetralogia de Fallot está dependente do grau de obstáculo da câmara de saída do ventrículo direito.
Se o obstáculo for ligeiro, as manifestações ocorrem entre as quatro e as seis semanas de vida com clínica de insuficiência cardíaca por hiperfluxo pulmonar, com taquipneia, recusa alimentar e má progressão ponderal, e sem cianose. Assemelha-se ao quadro clínico de uma comunicação interventricular de grandes dimensões.
Nos casos com obstáculo moderado, os recém-nascidos estão acianóticos e a doença é diagnosticada no decurso da investigação de sopro sistólico detectado nas primeiras semanas de vida. A cianose surge normalmente entre os seis e dezoito meses de idade, à medida que aumenta o obstáculo. Inicialmente intermitente, manifestando-se apenas durante o choro, mamadas ou outra actividade física, torna-se persistente e associada a cansaço e/ou lipotímia.
Os recém-nascidos com obstáculo grave apresentam, logo nos primeiros dias de vida, cianose que se agrava com o encerramento do canal arterial; por vezes esta forma é canal dependente e requer infusão de prostaglandinas, para manter a permeabilidade do canal arterial.
Dependendo da gravidade da doença, pode haver: cianose, atraso da progressão estaturo-ponderal, hipocratismo digital ou taquipneia. A cianose pode ser evidente apenas nas mucosas, ou estar ausente (“mascarada”) pela existência de anemia. O hipocratismo digital – que consiste no alargamento e aumento de espessura das extremidades dos dedos (dedos em “baqueta de tambor”), acompanhado do aumento da convexidade das unhas (unhas em “vidro de relógio”) – só é evidente quando há hipoxémia significativa e mantida; é raro o seu aparecimento antes dos seis meses de idade.
Na auscultação cardíaca o primeiro ruído é normal, e o segundo ruído é geralmente único. Pode auscultar-se estalido de expulsão aórtica (por dilatação da aorta ascendente). Geralmente ausculta-se sopro de ejecção, crescendo – decrescendo, tonalidade rude, mais audível no terceiro espaço intercostal junto do bordo esquerdo do esterno, com irradiação posterior, para o dorso. O sopro tem origem na zona de estenose infundibular (não na comunicação interventricular), e a sua intensidade e duração são inversamente proporcionais ao grau de estenose. Nas formas ligeiras o sopro é intenso e longo; nas estenoses moderadas é em crescendo – decrescendo; e, nas formas graves, é menos intenso e curto, podendo mesmo desaparecer durante episódios de hipóxia. Os sopros contínuos são audíveis na presença de persistência do canal arterial ou colaterais sistémico-pulmonares.
Exames complementares
Radiografia do tórax
A silhueta cardíaca evidencia as seguintes características: dimensões normais, concavidade do arco pulmonar, ponta levantada (coração em forma de bota), e diminuição da vascularização pulmonar. (Figura 1)
Nas crianças acianóticas a radiografia pode ser normal. Um arco aórtico direito pode ser visualizado em 25% dos doentes.
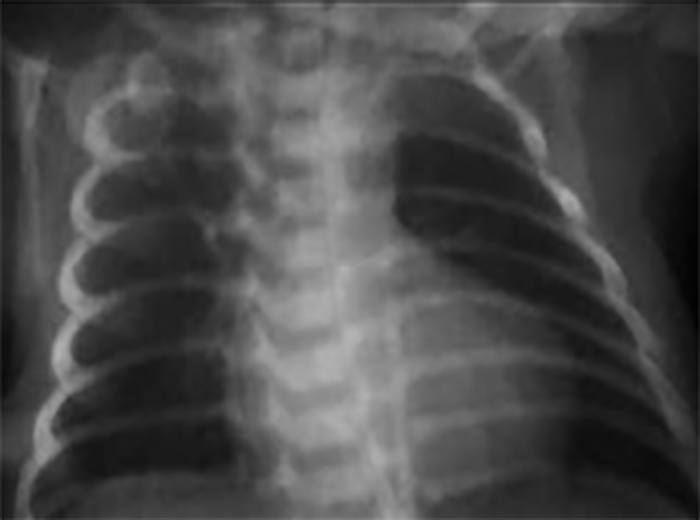
FIGURA 1. Radiografia de tórax (póstero-anterior) evidenciando: silhueta cardíaca com ponta levantada, compatível com hipertrofia ventricular direita, reentrância do arco pulmonar, e diminuição da vascularização pulmonar (tetralogia de Fallot)
Electrocardiograma
Os achados mais comuns são o desvio direito do eixo de QRS (entre +90º e +150º) e hipertrofia ventricular direita, com ondas R dominantes em V4R e V1, transição brusca de V1 para V2 e ondas S dominantes em V6. (Figura 2)
Nos casos de obstrução grave infundibular verifica-se uma transição brusca mais precoce, de V3R para V4R ou V4R para V1
Ecocardiograma
O ecocardiograma bidimensional é, actualmente, o método diagnóstico de eleição para avaliação de doentes com tetralogia de Fallot, permitindo avaliar as características anatómicas e funcionais (comunicação interventricular, câmara de saída do ventrículo direito, válvula pulmonar, tronco e ramos da artéria pulmonar) e, também, identificar lesões associadas. O estudo com Döppler quantifica o gradiente de pressão na zona do obstáculo pulmonar. (Figura 3)
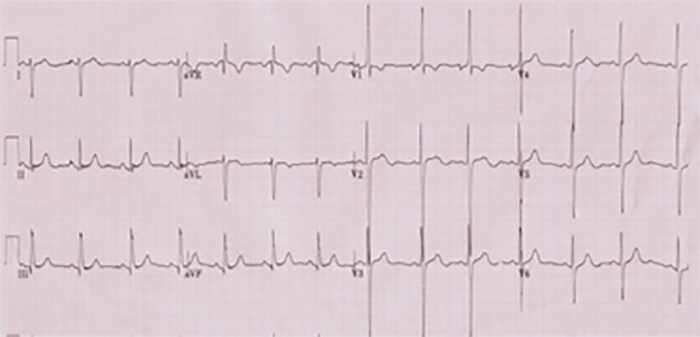
FIGURA 2. Electrocardiograma na tetralogia de Fallot: ritmo sinusal, eixo QRS no quadrante inferior direito e sinais de hipertrofia ventricular direita com transição brusca dos complexos ventriculares em V1-V2
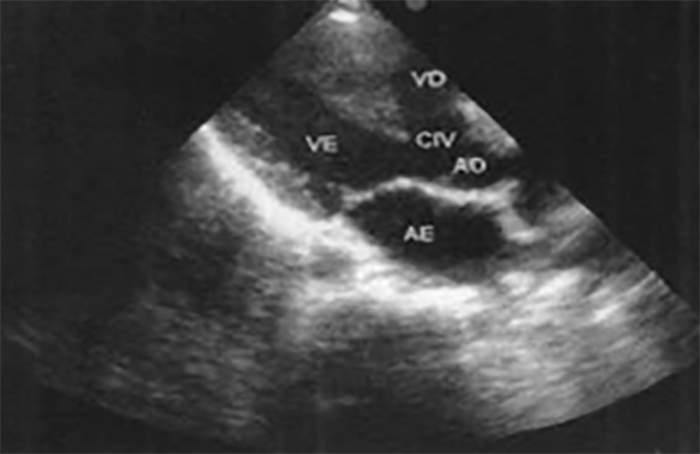
FIGURA 3. Ecocardiografia de tetralogia de Fallot em incidência parasternal; eixo longo: comunicação interventricular (CIV) com cavalgamento da aorta (AO) sobre o septo interventricular de 40%. (AE: aurícula esquerda; VE: ventrículo esquerdo; VD: ventrículo direito)
Cateterismo cardíaco
O cateterismo cardíaco diagnóstico encontra-se reservado para casos específicos: – nos quais o estudo ecocardiográfico não permita obter dados definitivos ou; – nos quais exista discrepância entre a anatomia e a evolução clínica encontrada. É sobretudo útil para uma caracterização pré-operatória detalhada da árvore pulmonar arterial, das anomalias das artérias coronárias e das colaterais sistémico-pulmonares. (Figura 4)
O cateterismo de intervenção é utilizado em recém-nascidos e lactentes com obstáculo pulmonar a múltiplos níveis (infundibular e valvular pulmonar) e cianose. Nestes doentes, a dilatação percutânea paliativa da válvula pulmonar permite melhorar o fluxo pulmonar, com a finalidade de evitar a realização de anastomose sistémico-pulmonar cirúrgica e promover o crescimento do anel e dos ramos da artéria pulmonar, diminuindo, assim, a necessidade de alargamento cirúrgico do anel com remendo transanular.
Ressonância magnética
A ressonância magnética cardíaca é actualmente um método fundamental no seguimento a longo prazo de doentes com tetralogia de Fallot corrigida cirurgicamente, especialmente em adolescentes e adultos. Permite uma avaliação anatómica e funcional detalhada destes doentes, nomeadamente quantificação das dimensões e função biventricular, detecção de regiões aneurismáticas ou acinéticas do ventrículo direito e sua função contráctil, avaliação dos ramos da artéria pulmonar, documentação de lesões residuais, nomeadamente estenoses e CIV residuais, e quantificação do grau de regurgitação pulmonar pós-operatório, de dilatação da raiz da aorta e do grau de fibrose.
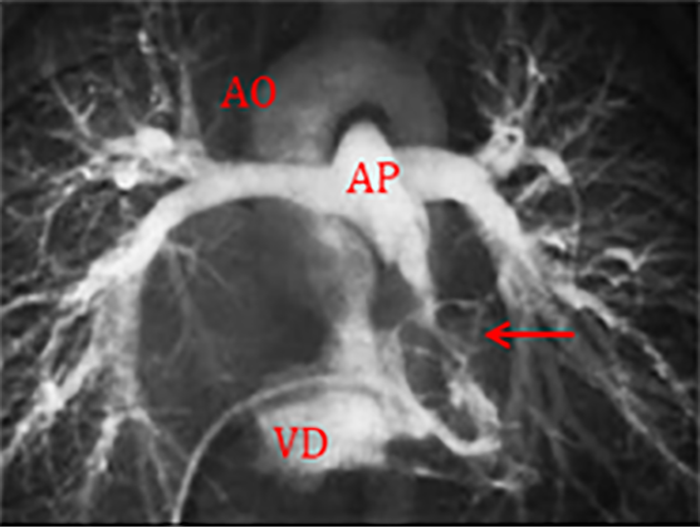
FIGURA 4. Ventriculografia direita em tetralogia de Fallot – visualiza-se em simultâneo: aorta (AO), artéria pulmonar (AP), obstáculo da câmara de saída do ventrículo direito (infundibular – seta). Boa anatomia dos ramos da artéria pulmonar
Tratamento
O tratamento dos doentes com tetralogia de Fallot (TOF) engloba o tratamento médico inicial, procedimentos cirúrgicos paliativos e de reparação intracardíaca, e o tratamento das complicações pós-operatórias.
Tratamento médico
A maioria dos doentes não necessita de tratamento no período neonatal. A excepção é constituída pelos casos com obstáculo pulmonar grave e oligoémia, e que se apresentam com cianose grave no período neonatal, após encerramento do canal arterial. Em tais circunstâncias, é necessária a utilização de prostagladinas de modo a promover a permeabilidade do canal arterial e a assegurar fluxo pulmonar, até realização de cirurgia precoce.
Em lactentes assintomáticos, acianóticos ou com cianose ligeira está recomendada a medicação com propranolol oral (dose 1-4 mg/kg/dia de 6/6h), com início aos três meses de idade, ou antes se agravamento do obstáculo pulmonar, com vista a prevenir o espasmo infundibular e o desenvolvimento de crises de hipóxia. É ainda recomendada a suplementação com ferro e multivitaminas, de modo a prevenir anemia, nomeadamente ferropénica. Os valores hematológicos devem ser avaliados periodicamente e a anemia ferropénica tratada. Valores normais de hemoglobina ou hematócrito numa criança cianótica indicam anemia.
As crises de hipóxia devem ser encaradas, como foi referido, como emergências. A criança deve ser colocada na posição genupeitoral com a cabeça mais baixa que os membros inferiores, e mantida em ambiente pouco aquecido. Estas medidas têm como objectivo aumentar a resistência vascular sistémica, o que diminuirá o shunt direito-esquerdo.
Deve ser administrado oxigénio, pelo seu efeito vasodilatador pulmonar e vasoconstritor sistémico. Na falência das medidas anteriores, deve ser administrado sulfato de morfina (0,1 mg/kg/dose por via subcutânea, intramuscular ou endovenosa) e expansão com volume (10-20 ml/kg, com soro fisiológico). O mecanismo de acção da morfina, desconhecido, resulta provavelmente da sua acção inotrópica e cronotrópica negativas, pelo seu efeito depressor do centro respiratório, e por quebrar o ciclo de hipóxia e agitação. O bolus de volume melhora o fluxo pulmonar anterógrado por aumento da pré-carga do ventrículo direito.
Deve ainda ser feita correcção da acidose metabólica com bicarbonato de sódio (1 mEq/kg).
A administração de propranolol (0,01 mg/kg por via intramuscular ou endovenosa) deve ser efectuada em unidades de cuidados intensivos. O propranolol exerce a sua acção através de um mecanismo cronotrópico e inotrópico negativos, com relaxamento da câmara de saída do ventrículo direito e melhoria do fluxo sanguíneo pulmonar. Se esta acção for insuficiente, a pós-carga sistémica pode ser aumentada através da administração de fenilefrina por via intravenosa (5 a 20 mcg/kg por dose). O propranolol por via oral (1-5 mg/kg/dia de 6/6 h) deve ser utilizado para profilaxia de novas crises. Depois de se tratar a crise de hipóxia e de se estabilizar o doente, deve promover-se o seu tratamento cirúrgico urgente tendo em atenção que, quando todas as medidas anteriores falham, o doente deve ser proposto para cirurgia cardíaca correctiva ou paliativa emergentes.
Todos os doentes (operados com lesões residuais ou não operados) devem ser submetidos a profilaxia da endocardite infecciosa.
Tratamento cirúrgico
O tratamento da tetralogia de Fallot é cirúrgico electivo, devendo ser realizado no primeiro ano de vida, preferencialmente nos primeiros seis meses de vida. Em função das capacidades técnicas dos centros, o tratamento tem-se realizado cada vez mais precocemente. A policitemia e as crises de hipóxia são indicações para tratamento cirúrgico mais precoce.
Classicamente os doentes eram submetidos primariamente a cirurgia paliativa, com o objectivo de aumentar o fluxo pulmonar e diminuir o grau de cianose.
Actualmente, a cirurgia paliativa está indicada apenas nos casos: – graves de tetralogia de Fallot, com hipoplasia dos ramos da artéria pulmonar; – que apresentem contraindicação para cirurgia cardíaca correctiva sob circulação extracorporal, nomeadamente por prematuridade, baixo peso; – que apresentem anatomia desfavorável. Constitui uma emergência nos lactentes com crises de hipóxia não controláveis com terapêutica médica.
Utiliza-se a anastomose de Blalock-Taussig, modificada, entre a artéria subclávia e os ramos da artéria pulmonar com interposição de um tubo de Gore-Tex®. Mais recentemente tem-se optado pela construção de um shunt central com interposição de tubo de Gore-Tex 3,5 mm, que apresenta como vantagens evitar distorções dos ramos das artérias pulmonares e complicações ortopédicas das toracotomias.
A cirurgia de correcção anatómica tem como objectivos: – desobstruir a câmara de saída do ventrículo direito; – separar a circulação sistémica da pulmonar; – preservar a função ventricular direita; – minorar o grau de regurgitação pulmonar pós-operatória.
Consiste no encerramento da comunicação interventricular com remendo, separando, assim, as circulações sistémica e pulmonar, e alargamento da câmara de saída do ventrículo direito, desobstruindo assim o fluxo pulmonar. O alargamento da câmara de saída do ventrículo direito é realizado através de valvulotomia pulmonar, ressecção de bandas musculares infundibulares e septo-parietais e, se necessário, alargamento do anel pulmonar com colocação de remendo de homoenxerto com válvula pulmonar mono cusp.*
*Válvula mono cusp significa que a substituição da válvula do doente é feita utilizando um enxerto valvular em que existe uma cúspide valvular com a finalidade de evitar insuficiência valvular. |
Prognóstico
O prognóstico a curto-médio prazo é excelente para lactentes e recém-nascidos submetidos a cirurgia cardíaca correctiva, com uma taxa de mortalidade perioperatória de 0 a 3%. As lesões residuais (obstrução da câmara de saída do ventrículo direito e/ou regurgitação pulmonar) de grau ligeiro são bem toleradas. Numa pequena percentagem de doentes poderá haver necessidade de reoperação para corrigir comunicações interventriculares residuais ou obstáculo pulmonar evolutivo.
O prognóstico a médio prazo dos doentes com tetralogia de Fallot é igualmente favorável, com uma taxa de sobrevivência de 90% aos 30 anos. É, contudo, expectável a necessidade de múltiplas intervenções terapêuticas ao longo da vida, e uma esperança média de vida inferior à da população geral. Os doentes com tal patologia devem manter seguimento regular pelo risco de complicações tardias, presentes em 15% dos doentes aos 20 anos de seguimento. No que respeita a complicações, as mais frequentes incluem: – o desenvolvimento progressivo de regurgitação pulmonar associada a dilatação e disfunção ventricular direita; – obstáculos residuais da câmara de saída do ventrículo direito, com necessidade de reintervenção cirúrgica ou percutânea; – dilatação da raiz da aorta; e – arritmias e morte súbita cardíaca.
As alterações do ritmo cardíaco são as complicações tardias mais frequentes após a cirurgia; na maioria das vezes correspondem a bloqueio de ramo direito, taquicardias auriculares e ventriculares, com risco aumentado de morte súbita cardíaca.
A maioria dos doentes fica assintomática, sem necessidade de terapêutica e com razoável tolerância ao exercício. Na ausência de lesões residuais significativas a gravidez é bem tolerada.
BIBLIOGRAFIA
Freitas I, Nogueira G, Kaku S. Tetralogia de Fallot. In Soares Costa JTS e Kaku S (eds). Cardiopatias Congénitas. Lisboa: Permanyer Portugal, 2005; 91-96
Garson AJr, Bricker JT, Fisher DJ, Neish SR (eds). The Science and Practice of Pediatric Cardiology. Baltimore: Williams & Wilkins, 1998
Guntheroth WG, Morgan BC, Mullins GL K. Physiologic studies of paroxysmal hyperpnea in cyanotic congenital heart disease. Circulation 1965; 31: 70-76
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Park MK (ed). Park’s Pediatric Cardiology for practitioners. Philadelphia: Elsevier, 2014
Schultz AH, Wernousky G. Late outcomes in patients with surgically treated heart disease. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2005; 8: 145-156
Zhu L, Yue JW, Yang XH. Target sequencing identifies a novel mutation of ZFPM2 in a Chinese newborn with tetralogy of Fallot. Ann Clin Lab Sci 2019; 49: 274-275