Introdução às anomalias congénitas do SNC
Os defeitos congénitos do SNC no RN, com uma frequência oscilando entre 0,5 e 5%, e representando cerca de 10% a 20% do total dos referidos defeitos, são responsáveis por 70% das mortes fetais e 40% das mortes durante o primeiro ano de vida.
Tais afecções integram um conjunto de entidades clínicas que podem ser sintetizadas do seguinte modo:
- Defeitos do tubo neural ou disrafismo
- Defeitos da migração, proliferação e diferenciação neuronais
- Anomalias da segmentação e da divisão cerebrais
- Hidranencefalia, porencefalia e quistos intracerebrais
- Defeitos congénitos do cerebelo
- Hidrocefalias
- Craniossinostoses ou craniostenoses
- Síndroma de Klippel-Feil
Neste capítulo é dada especial ênfase às situações que integram a alínea 1).
Noutros capítulos, são abordados temas referentes às restantes alíneas.
Sistematização e importância do problema
Os defeitos do tubo neural (DTN) (ou disrafismo) incluem anomalias congénitas da coluna e do cérebro; os mais frequentes são a spina bifida (SB) e a anencefalia (esta incompatível com a vida).
A spina bifida (SB) consiste no não encerramento do arco posterior de algumas vértebras, com possibilidade de herniação do tecido neural. (Figura 1).
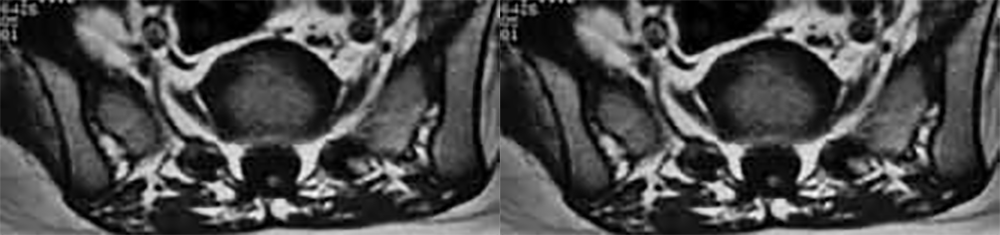
FIGURA 1. Imagens axiais de RM da coluna que mostram o não encerramento posterior das vértebras e a existência de hidro-siringomielia (seta). (ver adiante)
O espectro clínico dos DTN inclui ainda o encefalocele, a craniorraquisquise (anencefalia associada a raquisquise ou fissura congénita da coluna vertebral com exposição do tecido neural) e a iniencefalia (disrafismo na região occipital, acompanhado por retroflexão acentuada do pescoço e tronco).
Tais defeitos devem-se a um desenvolvimento anómalo do neuroporo durante a embriogénese, com disrupção do osso e das estruturas mesenquimatosas. A lesão primária neurológica vai afectar outros sistemas além do sistema nervoso, o que torna os DTN as anomalias de desenvolvimento mais complexas.
Tratando-se de multideficiências (coexistência de duas ou mais perturbações nas áreas motora, sensorial e cognitiva) de baixa incidência e prevalência que entram no âmbito das doenças raras, as mesmas obrigam ao recurso a cuidados de saúde de nível terciário dada a multiplicidade, especificidade e complexidade dos problemas habitualmente associados.
A SB constitui o paradigma de problema complexo implicando enorme consumo em recursos de saúde, com múltiplas consultas, tratamentos, internamentos e intervenções cirúrgicas não só neurológicas mas também ortopédicas e nefro-urológicas. A possibilidade de prevenção de muitas complicações, com melhoria significativa da qualidade de vida e redução substancial dos custos, passa necessariamente pelo ensino e crescente corresponsabilização do doente e família na prestação de cuidados.
Os doentes com SB têm compromisso motor e sensitivo, malformações ortopédicas, ausência de controlo de esfíncteres, e complicações renais secundárias à bexiga neurogénica.
Existem também complicações da hidrocefalia consequente, traduzidas frequentemente por dificuldades de aprendizagem, atraso mental, perturbações do equilíbrio, da marcha e problemas oftalmológicos. (ver adiante)
Devido à multiplicidade e complexidade dos problemas destes doentes, foi sentida a necessidade de se constituirem equipas multidisciplinares que pudessem prestar cuidados de saúde abrangentes e coordenados. No Hospital Dona Estefânia funciona desde 1985 um Núcleo de Spina Bífida onde são seguidas regularmente cerca de 220 crianças e adolescentes.
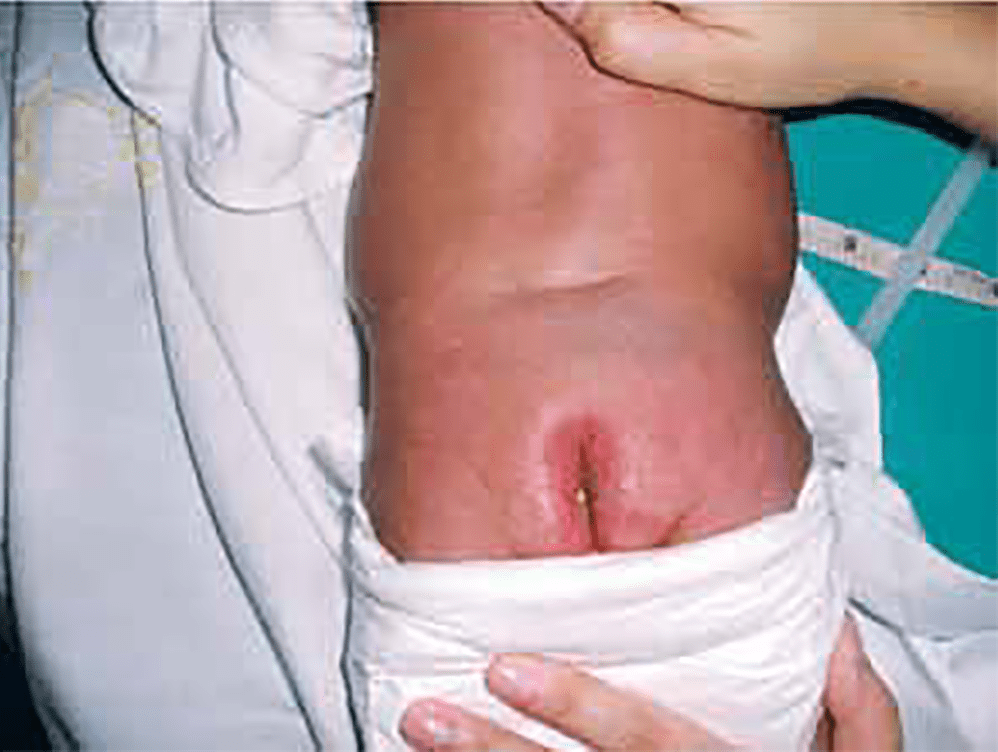
FIGURA 2. RN com uma forma fechada de SB (lipomielomeningocele). Nota-se tumefacção lombar, angioma cutâneo e fosseta mediana horizontal
Na prática utiliza-se o termo de spina bifida (SB) ou espinha bífida como sinónimo de DTN, atendendo a que as crianças com os outros tipos de DTN raramente sobrevivem. Esta anomalia localiza-se mais frequentemente na região lombo-sagrada, embora possa aparecer ao longo de toda a coluna. Compreende as formas fechadas e as formas abertas, consoante o tecido neural se encontra ou não coberto pela pele normal.
As formas fechadas podem incluir uma massa subcutânea (lipomielomeningocele, lipomeningocele, mielocistocele), que faz saliência na região lombo-sagrada (Figura 2).
As formas fechadas sem massa subcutânea compreendem o filum terminal ancorado, o lipoma intradural, o sinus dérmico ou mesmo disrafismos mais complexos como o quisto neuroentérico, a diastematomielia ou a agenésia caudal.
A chamada spina bifida oculta, que se encontra em 5% da população, diz respeito em sentido estrito apenas ao defeito ósseo do não encerramento de uma ou duas vértebras na transição lombo-sagrada (L5 e/ou S1), demonstrada nas radiografias desta zona, com completa integridade da medula e meninges; de referir que isoladamente não tem repercussão clínica.
As formas fechadas associam-se, por vezes, a alterações cutâneas (hipertricose, hemangiomas capilares, fossetas) na linha média da região lombo-sagrada (Figura 2 e 3).

FIGURA 3. SB oculta com um tufo de pêlos sinalizando o encerramento incompleto do arco posterior da vértebra. A medula e as meninges estão intactas

FIGURA 4. Meningocele com herniação das meninges através do defeito ósseo. Medula íntegra

FIGURA 5. Mielomeningocele. A medula e as meninges herniam através da abertura óssea
Das formas abertas faz parte o meningocele (Figura 4), em que já existe herniação das meninges através do defeito ósseo e que implica correcção neurocirúrgica; em geral, não se acompanha de qualquer sintomatologia motora. No mielomeningocele (Figura 5), a forma mais grave de SB (e aquela que habitualmente se subentende quando se faz referência a SB), existe procidência da medula espinal ou das raízes nervosas através do defeito ósseo, com lesões neurológicas mais ou menos importantes. Usa-se frequentemente o termo de SB quística para denominar o mielomeningocele e o meningocele. Em cerca de 80% dos casos a SB (na sua forma de mielomeningocele) acompanha-se de hidrocefalia (Figura 6) por malformação cerebral associada (malformação de Arnold Chiari II e/ou estenose do aqueduto de Sylvius).
Recordam-se, a propósito, as seguintes definições complementares:
- Hidrocefalia (ou hidrencefalia): dilatação das cavidades ventriculares e dos espaços subaracnoideus da cavidade craniana por pressão excessiva do LCR, (produzido pelos plexos coroideus nos ventrículos laterais) podendo determinar aumento do perímetro cefálico.
- Malformação de Arnold-Chiari: defeito congénito que consiste na descida do cerebelo e tronco cerebral para o canal vertebral e penetração das amígdalas cerebelosas no canal cervical com consequente hidrocefalia.
- Siringomielia: afecção crónica caracterizada pelo desenvolvimento progressivo, na medula cervical e cérvico-dorsal, de uma cavidade na substância cinzenta, atrás do canal ependimário, por obstrução da normal circulação de LCR ao nível do foramen magnum. Como consequência, surge atrofia muscular, sobretudo nos membros superiores com hipotonia, atrofia dos tegumentos e abolição da sensibilidade dolorosa e térmica. (Figura 7)
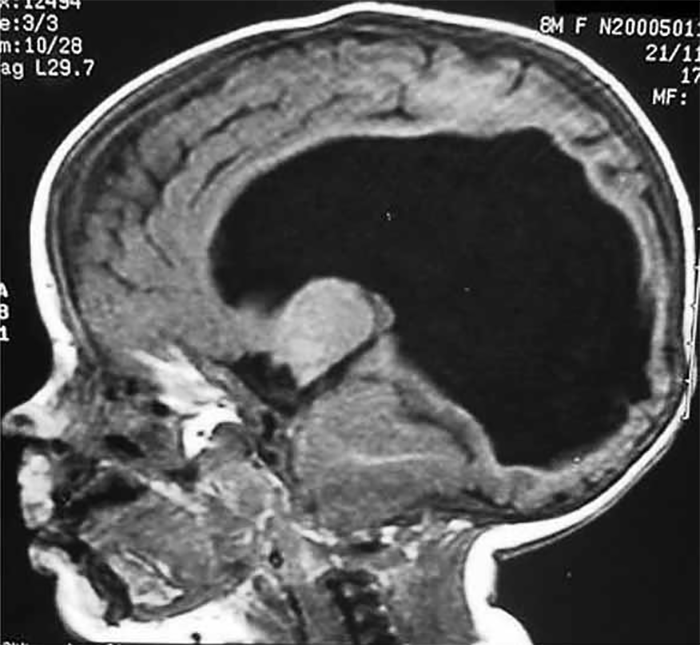
FIGURA 6. TAC evidenciando hidrocefalia com DVP em criança com mielomeningocele ao nível D12, siringomielia extensa, atrofia significativa do manto cortical, e atraso cognitivo grave
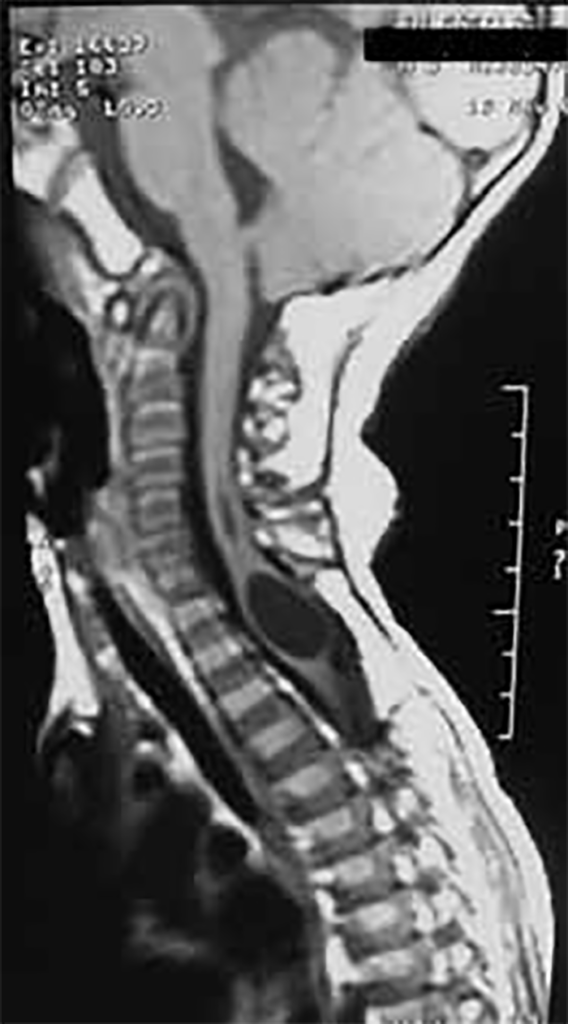
FIGURA 7. Siringomielia “septada” a nível cérvico-dorsal (com aspecto quístico), numa criança de três anos com mielomeningocele nível S1 e Arnold-Chiari II concomitantes
Aspectos epidemiológicos
A prevalência da SB tem vindo a descrescer nos países desenvolvidos, sendo actualmente cerca de 0,1/1000 nados-vivos. Isto deve-se, em parte, às possibilidades de diagnóstico pré-natal possibilitando a interrupção da gravidez nos casos mais graves; as ecografias pré-natais de alta resolução (ecografias morfológicas) permitem a visualização do defeito neural entre a 16ª e a 20ª semana de gestação em cerca de 99% dos casos. Reportando-nos ao estado nutricional, cabe referir que a ingestão de ácido fólico (4mg/dia) durante 3 meses antes da concepção e no 1ºtrimestre de gravidez em mulheres com antecedentes gravidez com DTN, diminui em cerca de 70% a recorrência de DTN (ver adiante).
Até à década de sessenta era escasso o número de doentes com SB que sobrevivia, pois não havia possibilidade de proceder a derivação da hidrocefalia. Logo que tiveram início as intervenções de derivação ventriculoperitoneal (DVP) a sobrevida foi aumentando e, actualmente, nos países ocidentais, cerca de 90% dos doentes atinge a idade adulta.
Etiopatogénese
A falência do encerramento do tubo neural ocorre nos estádios embrionários precoces da gastrulação e da neurulação (primeiras 6 semanas de gestação).
O defeito básico consiste no não encerramento primário do tubo neural, embora a reabertura secundária também seja considerada nalguns casos.
Os factores etiológicos podem ser exógenos (víricos, tóxicos, radioactivos, nutricionais, químicos, etc.) e endógenos (anomalias cromossómicas, ou génicas).
O momento da actuação da noxa sobre o feto é mais importante do que a própria natureza da noxa.
Nesta perspectiva cabe salientar o importante o papel desempenhado pela privação vitamínica na mãe, na data da fecundação, sobretudo de ácido fólico. Este aspecto explica o aumento de incidência da SB nas classes mais desfavorecidas e em situações de guerra ou de catástrofe, caracterizadas por carências nutricionais.
Relativamente aos factores genéticos, eles relacionam-se essencialmente com os genes, ainda não completamente identificados, que regulam o metabolismo do complexo folato-homocisteína, principal responsável pelo controlo dos mecanismos celulares de encerramento do tubo neural. O risco de uma mulher com um filho portador de SB vir a ter outro filho afectado é 20 vezes superior ao da população geral. No Núcleo de SB do HDE, houve 4 recorrências de fetos com SB em 165 mães de crianças afectadas, seguidas ao longo de 20 anos. O valproato de sódio e a carbamazepina, medicamentos antiepilépticos, aumentam a incidência de DTN quando tomados durante a gravidez, por muito provável interferência no metabolismo do ácido fólico. Na impossibilidade de mudar a terapêutica, estas mulheres devem obrigatoriamente receber suplementos de ácido fólico e, ao engravidarem, ser seguidas em consulta de alto risco, com ecografias obstétricas morfológicas de elevada resolução. Nas 136 crianças actualmente seguidas no Núcleo de SB do HDE, duas mães tinham tomado carbamazepina e uma valproato de sódio, durante a gravidez.
A solução de continuidade ao nível do tubo neural permite a excreção de substâncias produzidas no feto (alfa-feto-proteína [AFP], acetilcolinesterase) para o líquido amniótico, servindo de marcadores bioquímicos do defeito em causa. Por outro lado, o rastreio no soro materno de AFP entre as 16-18 semanas de gestação permite identificar fetos de risco.
A hidrocefalia, que surge na grande maioria dos casos de DTN, explica-se fundamentalmente por 3 mecanismos gerais:
- insuficiência de reabsorção do LCR pelas vilosidades aracnoideias de Pacchioni, sendo o mesmo segregado pelos plexos coroideus nos ventrículos laterais (por ex. trombose dos seios venosos);
- hipersecreção de LCR (raramente), por exemplo, por papiloma dos plexos coroideus;
- obstrução mecânica (98% dos casos) impedindo a circulação do LCR; para além de processos inflamatórios, cabe referir fundamentalmente tumores e anomalias congénitas já citadas antes, associadas a DTN, acrescentando a anomalia de Dandy-Walker (dilatação quística do IVº ventrículo por ausência congénita dos respectivos orifícios de evacuação – buracos de Magendie e Lushka) e atrésia do aqueduto de Sylvius.
A circulação do LCR depende dum gradiente de pressões; em situação normal a pressão intraventricular é ~ 180 mm H2O e a do seio longitudinal superior ~ 90 mm H2O.
A hidrocefalia que resulta de obstrução ao nível do sistema ventricular é chamada obstrutiva ou não comunicante; a que resulta de obliteração ao nível das cisternas subaracnoideias, ou de disfunção das vilosidades aracnoideias é chamada não obstrutiva ou comunicante.
Manifestações clínicas
Em termos de sistematização, os doentes com SB dividem-se em 3 grupos, de acordo com o nível da lesão: nível superior (L2 ou acima), nível médio (L3 a L5), e nível inferior (S1 ou abaixo). Quanto mais elevado for o nível da lesão (Figura 8) maior a probabilidade de ocorrência de hidrocefalia e maior o grau de incapacidade motora e de complicações secundárias.
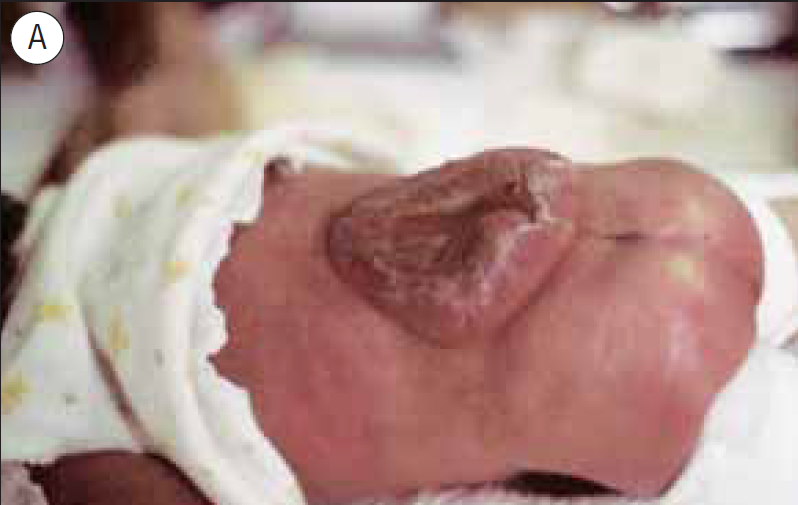
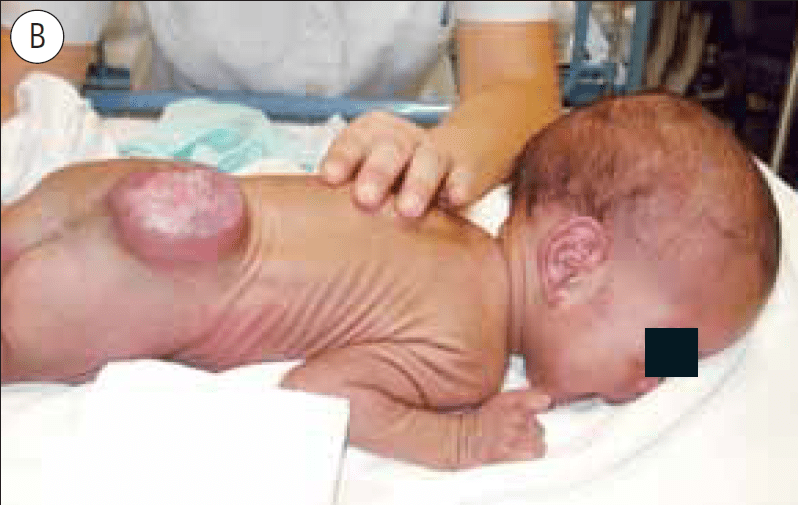
FIGURA 8. A: Mielomeningocele de nível superior (D10), com exposição do tecido neural. B: Criança vinda de África com um mês de vida. Mielomeningocele íntegro e hidrocefalia sintomática (vómitos e letargia)
Cerca de 40% dos doentes com SB desloca-se em cadeira de rodas. A lesão medular e/ou das raízes nervosas é responsável pela paraplegia mais ou menos grave, pelo compromisso da sensibilidade com risco de úlceras de pressão e queimaduras, pelas malformações e deformações ortopédicas, pela ausência de controlo dos esfíncteres vesical e anal, e pelas complicações nefro-urológicas.
A hidrocefalia que, como foi referido, ocorre na grande maioria dos casos de mielomeningocele, é a causa dos problemas cognitivos, visuais e de equilíbrio que alguns doentes com SB apresentam.
Nos doentes com SB sem hidrocefalia, a função cognitiva é habitualmente sobreponível à da população geral.
A epilepsia, presente num número restrito destes doentes (3% – na casuística do Núcleo do HDE), é habitualmente secundária a complicações da hidrocefalia.
Apneia, alteração de deglutição e estridor podem surgir nalguns doentes com SB, sobretudo lactentes, devido à malformação de Arnold-Chiari e a conflito de espaço a nível do foramen magnum, com disfunção dos pares cranianos inferiores.
Mais de metade dos doentes com malformação de Arnold-Chiari II apresenta siringo-hidromielia, logo nos primeiros anos de vida. Localizando-se habitualmente nas regiões cervical ou dorsal, traduz-se por compromisso das sensibilidades dolorosa e térmica nos dermátomos correspondentes, e diminuição de força e atrofia dos músculos da mão ou mesmo de todo o membro superior (Figura 7).
Praticamente todos os portadores de SB têm incontinência de esfíncteres urinário e anal, por compromisso do sistema nervoso autónomo. São frequentes as infecções urinárias, e cerca de um terço evoluirá para insuficiência renal se não houver um correcto acompanhamento tendo em conta as particularidades da bexiga neurogénica.
Com o crescimento existem sérias possibilidades de deterioração da marcha nas formas inicialmente ambulatórias, devido à baixa terminação da medula (L5-S1 em vez de L1 como nos indivíduos normais), e à sua fixação às estruturas envolventes (medula ancorada), com o consequente estiramento. Esta situação também é responsável pela deterioração nefro-urológica secundária ao agravamento da bexiga neurogénica, com pressão intravesical elevada, o que facilita o aparecimento de refluxo vesico-ureteral e de cicatrizes renais secundárias a infecção. Este problema neurocirúrgico deve ser atempadamente resolvido, logo que surjam os primeiros sinais neurológicos “de novo” (pés cavus, hiperreflexia, espasticidade distal com encurtamento do tendão de Aquiles, diminuição de força, atrofia dum dos membros inferiores ou síndromas álgicas) e/ou agravamento dos exames urodinâmicos, com repercussão clínica.
As deformações ortopédicas também são frequentes e determinadas pelo nível da lesão e complicações da medula ancorada. O pé equinovaro, a luxação da anca e a cifoscoliose são as alterações que motivam maior número de intervenções ortopédicas nesta população. As fracturas espontâneas nos membros inferiores ocorrem com frequência nas SB com nível mais elevado e maior compromisso motor; atendendo à ausência de sensibilidade nestes doentes, o diagnóstico pode ser tardio (Figura 9).
De salientar que cerca de 50% da população com SB tem hipersensibilidade ao látex.
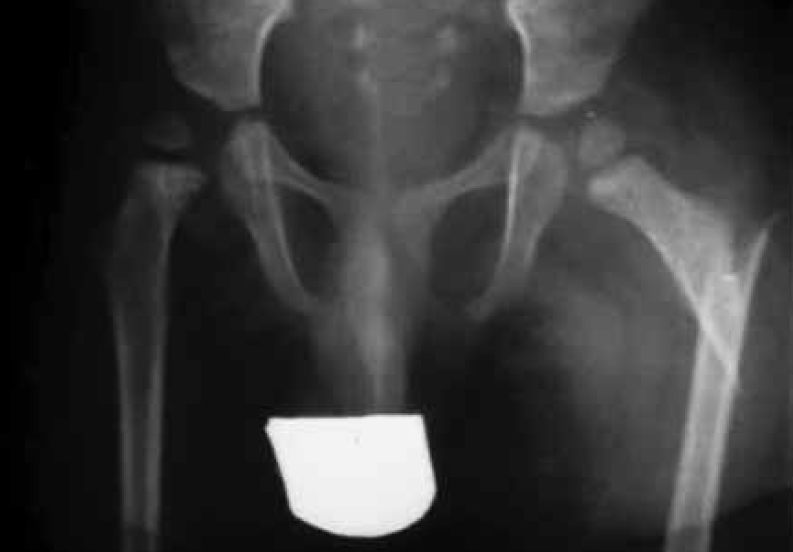
FIGURA 9. Fractura espontânea do colo do fémur esquerdo em criança com mielomeningocele nível L1
Diagnóstico
O diagnóstico do disrafismo espinhal pode ser feito a partir da 14ª semana de gestação através da ecografia pré-natal morfológica. A hidrocefalia na SB tem início, na maioria dos casos, no período pré-natal, a partir das 20 semanas de gestação. A RM fetal veio tornar possível uma melhor identificação dos DTN; o recurso a este exame é indispensável para fundamentar a decisão relativamente ao prosseguimento ou interrupção duma gravidez cursando com DTN.
Após o nascimento é ainda a RM o exame de escolha para uma adequada avaliação destas situações; sempre que possível deve ser realizada à totalidade do SNC antes do encerramento do DTN, para estudo evolutivo mais apurado.
A inexistência de pregas radiárias perianais ou a sua escassez apontam para o quadro de intestino neurogénico com maior probalidade de incontinência anal.
Nas formas fechadas deve ser sempre realizado o estudo imagiológico por RM da coluna, sobretudo se houver a associação de 2 ou mais sinais cutâneos.
A medula ancorada é uma complicação frequente das formas abertas e fechadas de disrafismo; pode tornar-se sintomática em qualquer idade, mas mais frequentemente na criança ou jovem adulto: desenvolvimento de sinais piramidais nos membros inferiores, deterioração da marcha, aumento da frequência de infecções urinárias, maiores dificuldades na continência, e desenvolvimento de escoliose.
Tratamento
O tratamento da SB já é possível iniciar-se durante a gravidez, com a cirurgia fetal. O encerramento do mielomeningocele (MM) in utero diminui a probabilidade de desenvolvimento de hidrocefalia, mas parece não melhorar muito a funcionalidade dos membros inferiores.
Trata-se duma área da cirurgia fetal, ainda em investigação, e restrita a alguns centros neurocirúrgicos; é, por isso, necessário avaliar mais estudos prospectivos comparando os resultados do encerramento no período pré-natal com os do encerramento no período pós-natal.
Após a criança nascer, o encerramento do mielo ou do meningocele deve realizar-se nas primeiras 24 a 72 horas de vida num bloco operatório isento de látex, medida que deverá ser sempre seguida em ulteriores intervenções cirúrgicas. Se houver rotura da membrana envolvente, a cirurgia deverá realizar-se logo nas primeiras horas de vida de modo a evitar a infecção e, assim, diminuir o risco de agravamento da lesão motora e o compromisso cognitivo. Quanto aos lipomeningoceles, a intervenção pode ser adiada vários meses ou mesmo anos, desde que não sejam muito volumosos e não apareçam sinais associados à medula ancorada.
A hidrocefalia pode estar presente desde o nascimento (em cerca de 15% dos mielomeningoceles) e a DVP pode realizar-se em simultâneo com o encerramento do MM. Na maioria dos casos a hidrocefalia torna-se aparente 2 a 3 semanas depois do encerramento do DTN. Daí a necessidade da derivação, colocando um tubo flexível no sistema ventricular cerebral (geralmente no ventrículo lateral direito) para drenar o excesso de LCR para o peritoneu. Nos raros casos em que não existe possibilidade de absorção pelo peritoneu, esta derivação deverá ser feita para uma das aurículas.
O “desancoramento” da medula melhora a disfunção da bexiga e evita a progressão de sinais piramidais nos membros inferiores. Uma vez estes instalados, já é problemática a sua regressão, embora se possa evitar a sua progressão.
A maioria das crianças com SB necessita de apoios para a sua mobilidade – talas, canadianas e/ou cadeiras de rodas. A obesidade é um dos problemas frequentes nesta população, a qual não só compromete ainda mais a sua deambulação, como aumenta o risco de doenças cardiovasculares.
A ausência de sensibilidade favorece o aparecimento de escaras, feridas ou queimaduras nas zonas afectadas devido à inexistência de dor. A sua cicatrização é lenta e obriga muitas vezes à imobilização prolongada e a longos internamentos hospitalares.
Outros factores condicionantes do prognóstico da SB, e causas frequentes de mortalidade, são as complicações (obstrução, infecção) das DVP para resolução da hidrocefalia. Mesmo as hidrocefalias sem válvula necessitam de vigilância periódica, pois existe sempre a possibilidade da sua descompensação, com repercussões a nível cognitivo visual e motor. No Quadro 1 figuram os sinais mais frequentes de disfunção duma DVP.
QUADRO 1 – Sinais de disfunção de DVP
Agudos
|
Insidiosos
|
Grande parte das crianças e jovens com SB tem alterações vesicais e intestinais (bexiga e intestino neurogénicos), implicando necessidade de aprendizagem do seu controlo e de tratamento de modo a obter, sempre que possível, uma continência social.
Para evitar lesões renais, os pais e mais tarde as próprias crianças (de preferência antes de entrarem na escola primária), devem aprender a fazer algaliações para esvaziar a bexiga de 4 em 4 horas, com pausa nocturna de 8 horas. A cateterização intermitente deve ser instituída logo nos primeiros meses de vida sempre que os resíduos urinários sejam superiores a 10% da capacidade vesical calculada para a idade da criança.
Prognóstico
São necessários exames complementares de diagnóstico seriados (urinoculturas, ecografias e cintigrafias renais, cistografias, estudos urodinâmicos, RM, TAC) e múltiplos tratamentos médicos e/ou cirúrgicos para prevenir e tratar as complicações ao longo da vida do doente com SB. A inexistência de equipas multidisciplinares que assegurem uma adequada vigilância do doente com SB reflecte-se habitualmente em deterioração da qualidade de vida.
Há menos de três décadas, poucos bebés com SB sobreviviam ao seu primeiro ano de vida. Hoje, graças a um melhor tratamento que passa, não só por uma sofisticação das técnicas actualmente disponíveis, mas também por um forte investimento na prevenção das complicações secundárias, em 90% dos casos é atingida a idade adulta.
Prevenção
O tubo neural desenvolve-se nas primeiras 4 semanas da gravidez, quando a maioria das mulheres ainda desconhece que está grávida. Reitera-se o que foi dito a propósito da importância do suplemento oral de ácido fólico na dose de 4 mg/dia (nos casos de antecedentes de SB em filho anterior), desde 1 mês antes da data planeada para a concepção e, pelo menos, ao longo do primeiro trimestre da gravidez, enquanto durar a neurulação; não se verificando antecedentes de risco, em idêntico período é recomendada a dose menor (0,4 mg).
As mulheres com epilepsia e que queiram engravidar devem evitar tomar o valproato de sódio ou a carbamazepina; caso não seja possível substituir esta medicação antiepiléptica, são imprescindíveis os suplementos pré e periconcepcionais com ácido fólico e a sua orientação para uma consulta de alto risco.
BIBLIOGRAFIA
Adzick NS, et al. Prenatal repair of myelomeningocele versus postnatal repair. N Engl J Med 2011; 364: 993 – 1004
Aicardi J. Diseases of the Nervous Central System. London: Mac Keith Press, 2009
Aslan AR, Kogan BA. Conservative management in neurogenic bladder dysfunction. Curr Opin Urol 2002; 12: 473-477
Blencowe H, Kancherla V, Moorthie S, et al. Estimates of global and regional prevalence of neural tube defects for 2015: a systematic analysis. Ann NY Acad Sci 2018; 1414:31-46. Doi: 10. 1111/nyas. 13548
Calado E, Loff C. The failures of spina bifida transdisciplinary care. Eur J Pediatr Surg 2002; 12: S51-52
Campagnoni AT, el al (eds). Developmental Neuroscience. Basel: Karger, 2008
Dias L. Orthopedic care in spina bifida: past, present and future. Dev Med Child Neurol 2004; 46: 579
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Guggisberg D, Hadj-Rabia S, Viney C, et al. Skin markers of occult spinal dysraphism in children. Arch Dermatol 2004; 140: 1109-1115
Kaufman BA. Neural tube defects. Pediatr Clin North Am 2004; 51: 389-419
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
McLone DG. Pediatric Neurosurgery. Philadelphia: Saunders, 2001
Moro M, Málaga S, Madero L (eds). Cruz tratado de Pediatria. Madrid: Panamericana, 2015
Roach ES (ed). Pediatric Neurology. Philadelphia: Elsevier, 2019
Van Dyke DC, Stumbo PJ, Berg MJ, Niebyl JR. Folic acid and prevention of birth defects. Dev Med Child Neurol 2002; 44: 426-429
Walsh DS, Adzick NS. Foetal surgery for spina bifida. Semin Neonatol 2003; 8: 197-205