Definições e importância do problema
A ataxia define-se como perturbação da coordenação dos movimentos voluntários. Pode manifestar-se na posição de pé (estática), durante a marcha (locomotora), ou durante a execução de um movimento (cinética). Trata-se de um problema relativamente comum em idade pediátrica; na sua forma de manifestação aguda não é raro que necessite de uma abordagem diagnóstica, pelo menos inicial, pelo pediatra ou pelo clínico geral no serviço de urgência.
Tratando-se dum tipo de alteração dos movimentos, importa, por razões didácticas, definir sucintamente outros tipos não abordados em capítulos específicos, mas integrando diversos problemas clínicos:
- coreia, como situação caracterizada por movimentos involuntários e irregulares, umas vezes rápidos, outras vezes lentos, acompanhada por hipotonia muscular e perturbação da coordenação (por exemplo coreia de Sydenham, coreia de Huntington);
- atetose, como movimentos involuntários, lentos e ondulantes, predominantes nas extremidades; tais movimentos são amplificados por emoções ou excitações, atenuados com o repouso e desaparecem com o sono;
- tremor, como sucessão de oscilações rítmicas involuntárias que agitam uma parte do corpo ou o corpo inteiro; podem ser contínuas ou intermitentes, rápidas ou lentas, discretas ou acentuadas;
- tique, como movimento anormal intermitente, súbito e involuntário, que resulta da contracção de um ou mais músculos; desaparece durante o sono e pode ser controlado temporariamente pela vontade; o blefarotique ou tique localizado nas pálpebras constitui um exemplo.
Aspectos semiológicos e nosológicos
A ataxia resulta duma disfunção do cerebelo ou das suas conexões. Com efeito, o cerebelo coordena os movimentos e os mecanismos de ajustamento postural e da marcha.
Para a compreensão dos problemas clínicos nas disfunções cerebelosas a analisar neste capítulo, será importante uma abordagem prévia de aspectos essenciais da fisiologia e da semiologia.
As lesões cerebelosas produzem um quadro clínico característico:
- incoordenação e tremor chamado intencional ou cinético; os movimentos de aproximação, por exemplo nas provas dedo-nariz e calcanhar-joelho, são afectados por um tremor (ou perturbação da amplitude dos movimentos) que se torna mais amplo com a aproximação do alvo (dismetria); os movimentos de perseguição ocular são afectados por oscilações lentas e oscilações rápidas em vez de se realizarem de uma maneira gradual e uniforme, impedindo a manutenção dos olhos numa posição excêntrica (nistagmo); o discurso é perturbado por uma perda de nitidez na articulação e por uma entoação variável que decompõe as palavras nos seus componentes silábicos: disartria cerebelosa que traduz perturbação motora dos órgãos de fonação;
- desequilíbrio, sem direcção predominante; o encerramento dos olhos pode agravá-lo ligeiramente, mas não se observa um verdadeiro sinal de Romberg como nas lesões vestibulares ou cordonais posteriores; a marcha tem uma base larga e os passos são irregulares na direcção e na amplitude;
- hipotonia, mais notória nas lesões agudas, estando os reflexos osteotendinosos preservados; nas lesões crónicas (degenerativas ou outras), a hipotonia é menor ou pode não se verificar.
Ou seja, a ataxia apresenta diversas expressões semiológicas tais como, disartria, dismetria, tremor intencional, nistagmo e ainda a diadococinésia (esta última traduzindo impossibilidade de execução rápida de movimentos alternantes como pronação-supinação). De acentuar que as marchas peculiares das miopatias, neuropatias e doenças vestibulares não constituem ataxias.
As ataxias integram um tópico muito extenso e complexo, sendo de referir os constantes avanços nos aspectos genéticos moleculares e terapêuticos, os quais têm dado origem a múltiplas classificações ao longo do tempo. Tal circunstância é, pois, susceptível de tornar as actualizações obsoletas a curto prazo.
Sob o ponto de vista genético existem formas com padrão autossómico dominante, autossómico recessivo, ligadas ao cromossoma X e de hereditariedade mitocondrial, salientando-se a quantidade cada vez maior de genes reconhecidos.
Existem grupos em que ao compromisso neurológico se somam sinais extraneurológicos ou sistémicos. Coadjuvando a clínica, a este respeito, salienta-se o papel orientador da RM na identificação de alterações anatómicas como aplasia, hipoplasia, atrofia cerebelosa, ou outras alterações da substância branca e cinzenta.
No que respeita às ataxias hereditárias, são considerados três grande grupos: congénitas, em geral não evolutivas, com início ou maior expressão na infância; degenerativas que são evolutivas; e ataxias intermitentes.
Neste capítulo são considerados, numa perspectiva clínica, três tipos de ataxia: aguda, recorrente e crónica.
Ataxia aguda
A ataxia aguda pode, por vezes, constituir um problema complexo de diagnóstico diferencial, o que releva a importância da história clínica. De facto, numa situação aguda é fundamental definir se se trata do primeiro episódio de afecção com antecentes familiares do mesmo foro, ou se existe a possibilidade de, entre outros aspectos, efeito tóxico.
Nesta alínea são abordadas as situações de ataxia aguda mais frequentes, devendo salientar-se as infecções e as intoxicações.
Ataxia aguda para ou pós-infecciosa
O quadro de uma criança com uma história de varicela recente, provavelmente ainda com um exantema característico desta situação que, ao acordar, recusa a posição de pé, manifesta um claro desequilíbrio e tem tremor, constitui um dos exemplos mais frequentes de ataxia aguda observados pelo médico no serviço de urgência.
A ataxia aguda pós-infecciosa por cerebelite é uma situação que parece ser devida a invasão directa de um agente infeccioso ou a uma resposta inflamatória mediada imunologicamente após uma infecção.
Agentes microbianos mais frequentemente implicados são: vírus varicela-zoster, Mycoplasma, o vírus de Epstein-Barr, o citomegalovirus e enterovirus. Deve ter-se em conta que em cerca de 30-50% dos casos não é identificável uma infecção associada ao quadro de ataxia, ou antecedendo-o.
O quadro clínico típico é o de uma criança entre os 2 e os 7 anos, que no decurso de uma doença exantemática ou outra doença infecciosa, ou cerca de 1 a 2 semanas depois, acorda, verificando-se ataxia que é habitualmente mais notória de início. A ataxia pode ser tão marcada que não permite a posição sentada e determina um tremor cefálico. Podem também coexistir nistagmo, disartria (existem raras descrições de mutismo), tremor intencional e dismetria nos 4 membros, simetricamente, com reflexos mantidos e com uma hipotonia ligeira global. Não se verifica depressão do estado de consciência, nem alteração major de comportamento. A recuperação começa habitualmente após 1 semana e é em geral completa, embora possa ser prolongada.
Uma história de ataxia aguda, com as características descritas, deve determinar, após a avaliação inicial, um período de observação em centro especializado e quase sempre (possivelmente com a excepção da ataxia associada a varicela, em que o diagnóstico é óbvio) a realização de um exame de imagem: pela ressonância magnética podem, por vezes, observar-se sinais de lesões cerebelosas hiperintensas.
A literatura não é consensual sobre a necessidade de realizar, face a um quadro de cerebelite aguda, um exame do LCR (pode encontrar-se neste contexto uma ligeira pleocitose linfocítica, sem outras alterações). A indicação para realizar punção lombar é essencialmente a de suspeita de um diagnóstico alternativo, como encefalite (depressão do estado de consciência, alteração de comportamento ou sinais neurológicos focais).
Intoxicação
É bastante frequente, entre crianças dos 1 a 5 anos, a intoxicação acidental. Muitos casos envolvem a ingestão de fármacos com um efeito sedativo que também causam ataxia e nistagmo (tranquilizantes, antidepressivos, antiepilépticos, anti-histamínicos, antitússicos). A ingestão intencional (mas oculta) de medicamentos deste tipo, ou das chamadas drogas de uso recreativo ou de álcool, são uma possibilidade a considerar em adolescentes. Nem sempre o rastreio laboratorial é positivo e, nestes casos, o diagnóstico poderá depender da evolução clínica ou da exclusão de diagnósticos alternativos.
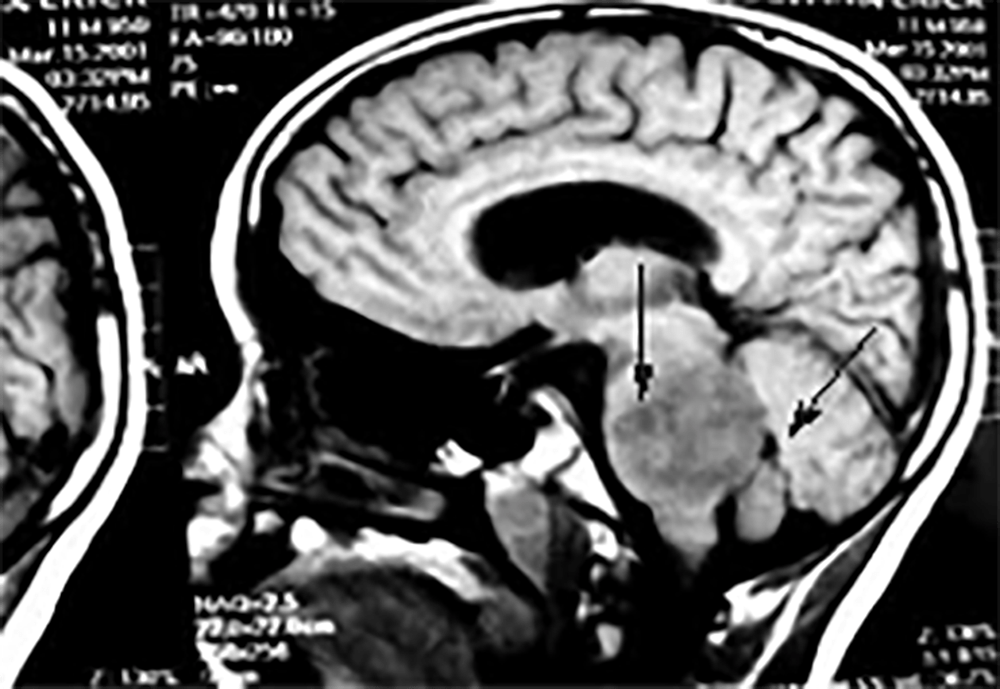
FIGURA 1. Exame de imagem (TAC) – Glioma da protuberância. Uma volumosa lesão assinalada na figura causa hidrocefalia por obstrução do IVº ventrículo. A história incluía cefaleias, vómitos, ataxia com um envolvimento do tronco e dos membros, e diplopia (por parésia do VIº par)
Tumores da fossa posterior
Uma criança com um tumor cerebral da fossa posterior pode recorrer ao serviço de urgência com uma história recente de ataxia (mais prolongada, semanas ou mesmo meses, eventualmente associada a cefaleias e/ou a vómitos). A ataxia não tem o carácter agudo descrito para a cerebelite e a história terá os elementos sugestivos de hipertensão intracraniana (cefaleias nocturnas que acordam o doente e no despertar, vómitos matinais que aliviam a cefaleia). O exame pode revelar uma ataxia de predomínio axial ou hemiataxia, eventualmente com sinais de compromisso de pares cranianos (mais frequentemente o VIº e o VIIº pares); e, na fundoscopia observar-se-á estase papilar. Mais frequentes neste contexto são o astrocitoma, o meduloblastoma, o ependimoma e o glioma da protuberância.
Os exames de imagem (TAC ou RM) permitem confirmar o diagnóstico (Figura 1).
Ataxia como sintoma de conversão
Perturbações do equilíbrio e da marcha, não raramente, podem surgir como sintomas de conversão em adolescentes. Nestes casos a marcha não tem uma base larga. A posição de “sentado” não causa qualquer dificuldade, mas a “de pé” manifesta-se por desequilíbrio, frequentemente espectacular; não se observam outras anomalias no exame neurológico.
Pseudo-ataxia epiléptica
Raramente uma criança com epilepsia de ausências ainda não diagnosticada, ou com certos tipos de epilepsia parcial refractária, ou ainda com epilepsia criptogénica, como a síndroma de Lennox-Gastaut, pode apresentar-se com um quadro clínico de status epiléptico não convulsivo: flutuação do estado de consciência, períodos sem contacto visual ou auditivo. Existe, contudo, possibilidade de realizar tarefas motoras de modo automático e, eventualmente, andar com algum desequilíbrio. Poderão também surgir mioclonias palpebrais e discretas clonias dos membros, multifocais, ou ainda episódios breves de nistagmo, sugerindo o diagnóstico de epilepsia.
O diagnóstico é confirmado pelo EEG. Estas situações exigem uma terapêutica antiepiléptica urgente, a decidir pelo neurologista pediátrico.
Enxaqueca da artéria basilar
A enxaqueca da artéria basilar, uma forma chamada «complicada» de enxaqueca, frequente na adolescência e no sexo feminino, pode incluir, como sintomas iniciais, de «aura», ataxia, nistagmo, vertigem, alterações visuais, parestesias e mesmo tetraparésia, que regridem já após o início de cefaleia occipital pulsátil (ver capítulo anterior).
O exame neurológico é normal após a recuperação e pode haver uma história precedente típica de enxaqueca (assim como antecedentes familiares de enxaqueca). Na abordagem inicial e na ausência dos referidos dados da história clínica, poderá ser necessário realizar um exame de imagem (para excluir lesão estrutural da fossa posterior), e EEG para excluir epilepsia occipital.
Ataxia pós-traumática
Em crianças, o sintoma pós-traumático mais frequente é a ataxia. A ataxia pós-traumática é habitualmente só axial, determinando um desequilíbrio. Provavelmente deve-se a uma perturbação transitória de funcionamento do tronco cerebral, o qual é submetido a trauma durante fenómenos de rotação e desaceleração contra a tenda do cerebelo.
Na fase aguda, após um traumatismo, uma criança com ataxia deve ser submetida a exame de imagem para excluir hemorragia na fossa posterior. É importante considerar também no diagnóstico diferencial a chamada concussão vestibular em que o desequilíbrio não é atáxico: em geral a criança recusa-se a mobilizar a cabeça, descreve uma sensação de vertigem e pode ter vómitos.
Opsoclónus-mioclónus
Trata-se duma situação rara em que uma criança com ataxia aguda, em geral com um importante componente mioclónico, tem associada uma considerável irritabilidade e uma anomalia oculomotora de tipo opsoclónus (movimentos conjugados, bruscos e amplos, involuntários dos olhos).
Este quadro clínico de opsoclónus-mioclónus pode ser pós-infeccioso ou paraneoplásico (neuroblastoma); dados recentes identificam-no como uma patologia autoimune do cerebelo.
Existe uma evolução crónica, frequentemente com flutuações; nos casos em que a etiopatogenia é paraneoplásica, persiste após o tratamento do tumor. Corticoterapia, gamaglobulina endovenosa e benzodiazepinas são as terapêuticas utilizadas.
Ataxia recorrente
A ataxia recorrente é menos frequente que a ataxia aguda, sendo o diagnóstico etiológico daquela muito diferente do anteriormente exposto para esta última.
Embora enxaqueca e epilepsia sejam doenças recorrentes, a ataxia como manifestação predominante daquelas não é comum, excepto nos já referidos status epiléptico não convulsante e na enxaqueca da artéria basilar.
A intoxicação acidental pode também ser recorrente, importando salientar que a síndroma de Munchausen «por procuração» é também, em função do contexto clínico e do ambiente em que vive a criança, uma causa a considerar.
No âmbito das ataxias recorrentes (as quais se devem, sobretudo, a doenças metabólicas e genéticas, faz-se referência apenas às seguintes situações:
- ataxia episódica (tipo 1 e 2)
- doença de Hartnup
- deficiência de PDHC
- leucinose (forma aguda intermitente)
A chamada doença vanishing white matter é uma encefalopatia relacionada com mutação no gene EIFB2A , localizado no cromossoma 3. Caracterizando-se por episódios recorrentes de alteração do estado de consciência, frequentemente após traumatismos cranianos minor ou doenças febris, evolui progressivamente para um défice neurológico de tipo atáxico e espástico, com uma relativa preservação das funções mentais. O estudo imagiológico pela RM cerebral permite identificar lesões da substância branca com a formação de quistos relacionáveis com hipomielinização.
Ataxia crónica
No diagnóstico de ataxia crónica devem ser consideradas separadamente: ataxia não progressiva e ataxia progressiva.
Ataxia congénita não progressiva
Neste grupo estão englobadas as ataxias congénitas devidas a defeitos congénitos do sistema nervoso. Existe discordância entre os achados de imagem e o quadro clínico: em geral, prevalece o défice cognitivo (como sintomatologia associada a um grave defeito de desenvolvimento do cerebelo) sobre os sinais clássicos de ataxia. Muitas destas crianças são hipotónicas mantendo reflexos osteotendinosos; algumas têm nistagmo ou estrabismo.
A situação mais caracterizada na literatura é a síndroma de Joubert (agenésia vermiana associada a um defeito de desenvolvimento do mesencéfalo, com atraso psicomotor, anormal controlo respiratório central com episódios de hiperventilação, nistagmo e displasia quística renal).
A hipoplasia congénita do cerebelo pode ser uni ou bilateral. Não são conhecidas as causas de hipoplasia unilateral, sendo de admitir que nalguns casos se trate de sequela atrófica de lesão pré-natal (provavelmente vascular ou infecciosa).
De salientar que o quadro de cerebelo hipoplásico associado a defeitos de migração neuronal de tipo polimicrogiria pode encontrar-se na infecção congénita por citomegalovírus e pode ocorrer associado a lissencefalia em mutações de genes da tubulina (TUBA1A).
Uma situação de ataxia com evolução subaguda implica a procura imediata de uma causa eventualmente tratável, como tumor da fossa posterior.
Ataxia crónica progressiva
Perante uma ataxia crónica progressiva é necessário pesquisar dados de história familiar (casos semelhantes na família sugerindo uma ataxia de tipo dominante e/ou consanguinidade, e irmãos afectados sugerindo uma doença recessiva).
Faz-se referência às seguintes entidades clínicas:
Abetalipoproteinémia
Uma história de ataxia com início na idade pré-escolar, arreflexia, nistagmo, precedida de atraso estaturoponderal e de síndroma de má absorção com esteatorreia, sugere abetalipoproteinémia. O diagnóstico é confirmado pelo achado laboratorial de níveis de colesterol e triglicéridos baixos, anemia de causa nutricional, e pela ausência de apolipoproteína B no plasma. O locus genético foi identificado no cromossoma 4 (gene MTTP: microsomal triglyceride transfer protein).
A terapêutica parentérica com vitamina E corrige as anomalias neurológicas na abetalipoproteinémia.
Ataxia-telangiectasia
A ataxia-telangiectasia é uma doença recessiva determinada por mutação no gene ATM (11q23.3), do que resulta uma proteína truncada não funcionante com efeitos diversos como hipersensibilidade a radiações ionizantes, atingimento do processo de reparação do ADN, inibição da sua síntese, incremento de rupturas cromossómicas, com consequentes anomalias imunológicas e incremento da apoptose.
Os sinais clínicos precoces são as infecções sinopulmonares recorrentes (frequentemente há défice de imunoglobulinas, mais frequentemente IgA associada a subclasses de IgG) e, por vezes, uma anomalia de movimento do tipo coreoatetose verificável nos primeiros dois anos de vida.
A ataxia surge subsequentemente e é progressiva. Na maioria dos doentes vem a desenvolver-se uma anomalia dos movimentos oculares chamada apraxia oculomotora (incapacidade de execução de movimentos voluntários coordenados apesar de se conservarem as funções musculares e sensoriais).
As telangiectasias (mais frequentes nas conjuntivas e nos pavilhões auriculares) observam-se após os dois anos. Na adolescência é frequente a perda da marcha autónoma pelo agravamento da ataxia e por neuropatia axonal progressiva.
Há um risco muito significativo de neoplasia, sobretudo linfoma e leucemia.
Quase todos os doentes têm níveis elevados de alfafetoproteína no soro e, em cerca de 80% dos casos, verifica-se deficiência de imunoglobulinas.
A RM-CE em fases avançadas evidencia atrofia cerebelosa.
O tratamento, que não é específico, baseia-se essencialmente na administração de imunoglobulina nos casos de infecções recorrentes, na fisioterapia e na terapia ocupacional. Salienta-se que é importante o diagnóstico precoce, a vigilância clínica atendendo à detecção de eventual surgimento de tumores, o conselho genético e o diagnóstico pré-natal com estudos moleculares.
Ataxia sem telangiectasia
Um fenótipo de «ataxia sem telangiectasia e sem imunodeficiência» foi descrito ao longo de vários anos, actualmente com 4 genes conhecidos – AOA1-AOA2-AOA3 e AOA4, destacando-se duas entidades: ataxia-telangiectasia-like disorder” e ataxia and oculomotor apraxia.
Ataxia de Friedreich
A ataxia de Friedreich é uma ataxia transmitida de modo recessivo; é explicada por uma mutação com expansão do gene denominado frataxina, no cromossoma 9. Está actualmente demostrado que a frataxina é uma proteína mitocondrial e que as mutações envolvidas nesta doença causam um défice desta proteína nas mitocôndrias e acumulação tóxica de ferro.
O quadro neurológico é o de ataxia progressiva que pode ter início entre os 2 e os 16 anos. A disartria é frequente e precoce. São típicos arreflexia e pés cavus, respostas plantares extensoras e hipostesia postural e vibratória. A perda da marcha autónoma ocorre cerca de 15 anos após o início dos sintomas.
Diabetes mellitus, cardiomiopatia hipertrófica e escoliose são as complicações não neurológicas mais frequentes.
Não há ainda um tratamento curativo para esta situação. O tratamento com antioxidantes como vitamina E e coenzima Q10 poderá retardar a progressão da doença.
Doenças mitocondriais
Um grupo importante de doenças, com expressão neurológica e sistémica que podem manifestar-se por ataxia progressiva, é constituído pelas doenças mitocondriais.
Os tecidos que exprimem clinicamente com mais frequência um defeito de função mitocondrial são aqueles que têm um maior consumo energético, nomeadamente o cérebro e o músculo.
As anomalias de função mitocondrial podem ter repercussão no ADN mitocondrial ou nuclear e podem ocorrer “de novo” ou ser herdadas. Devido a um fenómeno chamado heteroplasmia, os vários tecidos podem ser portadores, no mesmo indivíduo, de maior ou menor número de mitocôndrias com mutante, o que condiciona variantes do quadro clínico, da gravidade e da expressão nos diferentes membros de uma família.
Em geral deve suspeitar-se de doença mitocondrial perante um quadro clínico de doença neurológica, de ataxia, demência, neuropatia, miopatia, surdez, baixa estatura, diabetes, cardiomiopatia, independentemente dos antecedentes familiares (Parte XXXII).
A ataxia progressiva mais bem caracterizada em doenças mitocondriais ocorre nas síndromas de Kearns-Sayre (ataxia, retinopatia pigmentar, surdez, cardiopatia com bloqueio aurículo-ventricular) e NARP (neuropatia, ataxia, retinopatia pigmentar e surdez).
Outras ataxia crónicas
Além das situações referidas que frequentemente combinam ataxia com sinais piramidais e demência, várias outras doenças genéticas raras necessitam de ser consideradas no diagnóstico diferencial de uma ataxia progressiva infantil. Citam-se a doença de Nieman-Pick, as gangliosidoses juvenis (incluindo a doença de Tay-Sachs), a leucodistrofia metacromática, a doença de Krabbe juvenil, a adrenoleucodistrofia ligada ao cromossoma X, a doença de Refsum e a xantomatose cerebrotendinosa.
Ataxias de tipo dominante
Faz-se referência ainda a um grupo de ataxias com modo de transmissão dominante. O início dos sintomas é habitualmente na idade adulta; mas excepcionalmente pode ocorrer na infância ou na adolescência, por vezes com uma expressão clínica diferente. É o caso da doença de Machado-Joseph (SCA3: spinocerebellar ataxia type 3) e da SCA1 (spinocerebellar ataxia type 1).
AGRADECIMENTOS
À Dra. Leonor Bastos Gomes (Neurorradiologista do Hospital de Dona Estefânia) pela cedência da foto da Figura 1.
BIBLIOGRAFIA
Albin RL. Dominant ataxias and Friedreich ataxia: an update. Curr Opin Neurol 2003; 16: 507-514
Fenichel GM. Clinical Pediatric Neurology. Philadelphia: Saunders, 2001
Garone G, Reale A, Vanacore N, et al. Acute ataxia in paediatric emergency departments: a multicentre Italian study. Arch Dis Child 2019 Apr 4. pii: archdischild-2018-315487. doi: 10.1136/archdischild-2018-315487
Goldman L, Schafer AI (eds). Goldman-Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Jarman PR, Wood NW. Genetics of movement disorders and ataxia. J Neurol Neurosurg Psychiatry 2002; 73: 22-25
Jen JC, Graves TD, Hess EJ, et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007; 130; 2484 – 2493
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Luetje M, Kannikeswaran N, Arora R, et al. Utility of neuroimaging in children presenting to a pediatric emergency department with ataxia. Pediatr Emerg Care 2019 Mar 29. doi: 10.1097/PEC.0000000000001823.
Moro M, Málaga S, Madero L (eds). Cruz tratado de Pediatria. Madrid: Panamericana, 2015
Ryan MM, Engle EC. Acute ataxia in childhood. J Child Neurol 2003; 18: 309-316
Ruggieri VL, Arberas CL. Ataxias hereditarias. Rev Neurol 2000; 31: 288 – 296
Salman MS. The cerebellum: it’s about time! But timing is not everything- new insights into the role of the cerebellum in timing motor and cognitive tasks. J Child Neurol 2002; 17: 1-9
Tarsy D, Simon DK. Dystonia. NEJM 2006; 355: 818-829
Vedolin L, Gonzalez G, Souza CF, et al. Inherited cerebellar ataxia in childhood: a pattern-recognition approach using brein MRI. AJNR 2013; 34: 925 – 934