Definição
Na sequência do capítulo anterior, reitera-se que o crescimento humano é o resultado da expressão fenotípica duma potencialidade genética modulada pela nutrição, pela homeostase do meio interno, por certas hormonas e por múltiplos factores de crescimento. As alterações surgidas ao nível de um ou vários sistemas reguladores originam falência do padrão considerado normal, o qual se pode manifestar sob a forma de atraso ou aceleração do crescimento. Neste capítulo procede-se à abordagem sucinta das situações de atraso consubstanciando na prática o conceito de baixa estatura.
Nesta perspectiva, baixa estatura (BE) define-se auxologicamente por estatura inferior a dois desvios-padrão (DP) em relação à média correspondente para a idade, sexo e grupo populacional.
Etiopatogénese e classificação
Tratando-se dum problema complexo, pela observação do Quadro 1 discriminam-se múltiplos factores etiológicos envolvidos.
A BE idiopática define-se auxologicamente por estatura inferior a 2 DP em relação à média correspondente para a idade, sexo e grupo populacional, após exclusão de doença sistémica, endócrina, nutricional ou anomalias cromossómicas. Este critério engloba crianças com peso e comprimento normais ao nascer e sem défice de hormona do crescimento. Estima-se que 60-80% das crianças com BE cumpram os critérios de BE idiopática.
Tal conceito engloba um grupo heterogéneo de crianças sem causa identificada, incluindo subgrupo de crianças:
- Com baixa estatura familiar; e
- Com baixa estatura no contexto de atraso constitucional do crescimento e puberdade (ACCP).
QUADRO 1 – Baixa estatura – classificação etiopatogénica
| BE idiopática Variantes do normal | BE de causa conhecida Variantes patológicas do crescimento | |
Baixa estatura familiar Atraso constitucional do crescimento e puberdade Baixa estatura idiopática não familiar | Primária | Secundária |
Entidades clínicas de causa genética:
Osteocondrodisplasias congénitas: acondroplasia, hipocondroplasia Crianças pequenas para a idade gestacional: restrição de crescimento intrauterino, sem crescimento compensatório suficiente | Entidades clínicas de causa endócrina:
Metabólica: alterações do metabolismo dos lípidos, hidratos de carbono ou proteínas Doenças de órgão ou sistémicas: cardíaca, pulmonar (fibrose quística), hepática, intestinal (doença celíaca; síndroma de intestino curto), renal (insuficiência renal crónica), hematológica (anemia crónica), artrite Psicossocial: privação afetiva Iatrogénica: glucocorticóides; terapia anti-neoplásica (quimioterapia, irradiação do SNC) | |
O diagnóstico de uma variante do crescimento normal implica avaliação rigorosa e demonstração da integridade de todos os mecanismos de crescimento.
Avaliação clínica
A avaliação de uma criança com baixa estatura inclui a realização de anamnese pormenorizada e exame objetivo completo. No Quadro 2 salientam-se os dados mais relevantes a ter em conta e obrigatoriamente a inquirir. A presença de desproporção entre segmentos ou de sinais dismórficos poderão levar a admitir causas patológicas específicas, numa minoria dos casos. Os registos das avaliações prévias de somatometria são de extrema importância para se poder traçar a curva de crescimento, a qual constitui instrumento de avaliação essencial perante casos de BE.
QUADRO 2 – Baixa estatura – avaliação clínica
| Antecedentes familiares | · Estatura dos pais e irmãos (medição na consulta, se possível) · Cálculo da estatura-alvo familiar · Marcos de puberdade dos pais e irmãos (menarca, início de barba) |
| Antecedentes pessoais | · Gestação e parto (intercorrências) · Somatometria ao nascer (RCIU) · Problemas/anomalias congénitas detetadas durante o período neonatal · Neurodesenvolvimento |
| Antecedentes patológicos | · Doenças anteriores – infecções de repetição, diarreia crónica, cardiopatia, asma e seu tratamento (corticoterapia) |
| História social | · Eventos disruptivos na família. Privação afectiva |
| Dados antropométricos prévios | · Construção da curva de crescimento com base nos registos do BSIJ · Cálculo da velocidade de crescimento |
| Exame objectivo | · Peso, estatura · Relação peso/estatura · Proporção entre segmentos – tronco, membros superiores e inferiores · Desenvolvimento pubertário com especificação dos estádios · Pesquisa de dismorfias · Pressão arterial e frequência cardíaca · Palpação da glândula tiroideia |
A partir dos elementos recolhidos calculam-se:
- Estatura-alvo familiar – estimativa do potencial genético de altura da família, útil do ponto de vista comparativo. Calcula-se como uma média ponderada da altura dos pais, ajustada para o sexo da criança:
- Rapaz: [(altura da mãe + 13) + altura do pai] / 2
- Rapariga: [altura da mãe + (altura do pai-13)] / 2
Constitui critério de ulterior investigação a verificação de uma estatura inferior à estatura-alvo familiar em pelo menos 1,5 DP;
- Velocidade de crescimento (VC) – incremento em estatura por unidade de tempo, que se calcula e exprime em centímetros por ano (cm/ano) – ver capítulo anterior. O intervalo mínimo para poder ser calculada com rigor é de 6 meses.
Também, tal como referido no capítulo anterior, constitui critério para investigação, e sinal de alarme de causa patológica de BE, uma velocidade de crescimento inferior a 4 cm/ano, e/ou inferior ao P25 em curvas de velocidade de crescimento;
- Relação peso/estatura
- Diminuída – peso mais afetado do que a estatura: sugestivo de doença crónica ou desnutrição;
- Aumentada – estatura mais comprometida do que o peso: maior probabilidade de causa endocrinológica.
Diagnóstico diferencial e exames complementares
A avaliação da idade óssea (IO) através da radiografia do punho é um elemento da maior importância para o raciocínio clínico e diagnóstico diferencial, de forma integrada com os dados clínicos (Quadro 3).
QUADRO 3 – Baixa estatura – padrões de crescimento e diagnóstico diferencial
Abreviaturas: IO → idade óssea; IC → idade cronológica | ||
| IO versus IC | Velocidade de crescimento | Diagnóstico diferencial |
| IO ≅ IC | Normal | · Baixa estatura familiar · Síndromas de causa genética · Displasias ósseas · Restrição do crescimento intrauterino / RCIU |
| IO < IC | Normal | · Atraso constitucional do crescimento e puberdade · Doença crónica ou desnutrição ligeiras |
| IO << IC | Diminuída | · Causa endócrina · Doença crónica ou desnutrição graves |
Constituem sinais de alarme de causa patológica de baixa estatura, nomeadamente do tipo endócrino: estatura inferior a -3 DP, crescimento com velocidade inferior ao percentil 25, IO com atraso superior a 2 anos em relação à IC. Nestas situações está indicada ulterior investigação de eventual défice de GH.
Em todas as crianças com estatura inferior a -2 DP (ou abaixo do percentil 3), a avaliação clínica deve ser complementada com exames auxiliares de diagnóstico. Além dos exames dirigidos a eventual suspeita clínica de causa patológica, deve ser feita uma avaliação geral conforme sugerido no Quadro 4.
Se os resultados obtidos forem normais, poder-se-á, numa fase inicial, vigiar o crescimento da criança, monitorizando a velocidade de crescimento. Salienta-se que, pelo facto de a secreção de GH ocorrer de forma pulsátil, não está indicado o doseamento basal desta hormona, mas sim do factor de crescimento IGF-I e respectiva proteína transportadora IGFBP3, cuja concentração sérica não é afectada pela pulsatilidade.
QUADRO 4 – Baixa estatura – exames complementares de diagnóstico
| Exames imagiológicos | · Radiografia do punho – idade óssea |
| Exames laboratoriais gerais | · Hemograma completo · Marcadores de inflamação (velocidade de sedimentação, proteína C-reactiva / PCR) · Ureia, creatinina, ionograma · Gasometria · Cálcio, fósforo, magnésio, fosfatase alcalina · AST, ALT, GGT |
| Exames para avaliação de autoimunidade | · Autoanticorpos anti-transglutaminase IgA |
| Exames endocrinológicos | · Insulin-like growth factor I (IGF-I) e IGF-I Binding Protein 3 (IGFBP3) · Tirotropina (TSH) e tiroxina livre (T4L) |
| Exames de genética | · Cariótipo (se paciente do sexo feminino) |
Formas clínicas
Abordam-se em seguida as causas mais relevantes de baixa estatura na criança. As mais prevalentes integram o grupo designado por baixa estatura idiopática, constituindo variantes do crescimento normal (baixa estatura familiar e atraso constitucional do crescimento e puberdade). Entre as causas patológicas, o défice de hormona do crescimento, a síndroma de Turner e as crianças pequenas para a idade gestacional sem crescimento compensatório têm em comum a possibilidade de tratamento com hormona de crescimento biossintética.
1. BAIXA ESTATURA FAMILIAR
Trata-se de uma das causas mais frequentes de baixa estatura, que traduz a forte influência dos factores genéticos na estatura final de um indivíduo. Sendo um diagnóstico de exclusão, obriga ao seguimento continuado da criança ao longo do tempo, a fim de se detectar atempadamente qualquer desvio.
Uma criança com baixa estatura familiar nasce habitualmente com peso e comprimento adequados à idade gestacional, vindo a cruzar percentis de estatura durante os dois primeiros anos de vida, fase em que os fatores genéticos passam a ter maior influência sobre o crescimento infantil. Após estabilizar num percentil igual ou inferior ao 3, a criança cresce com velocidade de crescimento normal e apresenta uma maturação óssea adequada à idade cronológica. O pico de crescimento e maturação pubertária ocorrem na idade habitual.
2. ATRASO CONSTITUCIONAL DO CRESCIMENTO E PUBERTÁRIO
Também no diagnóstico desta entidade clínica os antecedentes familiares são de extrema importância, visto que se trata de uma situação de incidência familiar, cujas causas não estão completamente esclarecidas.
A somatometria ao nascer é habitualmente adequada à idade gestacional, e o crescimento é normal até ao início da idade pré-escolar, período a partir do qual poderá surgir desaceleração. A velocidade de crescimento volta a ser normal durante a infância, ocorrendo desaceleração nos anos pré-puberais, com cruzamento de percentis. Verifica-se nestes casos um atraso da maturação óssea e sexual.
O diagnóstico de exclusão de tal situação nem sempre é linear, exigindo avaliação complementar e monitorização clínica e auxológica, visto que o padrão de crescimento pode ser idêntico ao que ocorre em formas ligeiras de doença crónica (por exemplo doença de Crohn, doença celíaca, acidose tubular renal, etc.). O diagnóstico é confirmado quando é atingida a estatura-alvo familiar, após o surto de crescimento pubertário, que é tardio (pelos 14 anos nas raparigas e 16 anos nos rapazes), mas suficiente para atingir o normal potencial genético.
O Quadro 5 resume as principais características destas duas formas de baixa estatura idiopática. Em ambos os casos, por definição, o diagnóstico implica exclusão de doença sistémica, endócrina, nutricional ou anomalias cromossómicas.
QUADRO 5 – Baixa estatura familiar e atraso constitucional do crescimento e pubertário
Abreviaturas: IO → idade óssea; IC → idade cronológica; EAF → estatura alvo familiar | ||
| Baixa estatura familiar | Atraso constitucional do crescimento e da puberdade | |
| Antecedentes familiares | Familiares com baixa estatura | Atraso pubertário; surto de crescimento tardio; mãe com menarca tardia |
| Estatura alvo familiar (EAF) | Baixa | Normal |
| Somatometria ao nascer | Normal | Normal |
| Velocidade de crescimento | Normal | Diminuída nos primeiros 3-5 anos de vida e na fase pré-pubertária |
| IO versus IC | IO normal: IO @ IC | Atraso significativo de IO: IO < IC |
| Puberdade | Normal | Atraso pubertário |
| Estatura final | Correspondente a EAF (baixa) | Correspondente a EAF (normal) |
3. DÉFICE DA HORMONA DE CRESCIMENTO
Etiopatogénese
O défice de GH (hipopituitarismo) é uma causa rara de baixa estatura. Pode ser idiopático (na maior parte dos casos) ou de causa conhecida – congénita ou adquirida. As causas congénitas podem ser genéticas, associadas a defeitos estruturais do sistema nervoso central (displasia septo-óptica, agenésia do corpo caloso, síndroma da sela turca vazia) e/ou a defeitos da linha média (fenda palatina, incisivo central único).
As causas de défice de GH adquirido incluem os tumores do sistema nervoso central (craniofaringeoma, adenoma hipofisário), bem como as relacionadas com as respectivas intervenções terapêuticas (cirurgia, irradiação e/ou quimioterapia). Menos frequentemente, ocorrem também causas traumáticas ou infecciosas.
Manifestações clínicas
O quadro clínico do défice de GH é variável consoante a idade de apresentação.
No período neonatal acompanha-se de outros défices do eixo hipotálamo-hipofisário, traduzindo-se por hipoglicémia neonatal (défice de GH e ACTH / cortisol), micropénis (défice de gonadotrofinas) e icterícia neonatal prolongada. A díade de hipoglicémia neonatal e micropénis deve constituir um alerta clínico para o diagnóstico de hipopituitarismo, implicando tratamento urgente. Saliente-se que nestes casos o hipotiroidismo central não é detectado pelo rastreio neonatal do Programa Nacional de Diagnóstico Precoce, no qual são identificadas amostras com TSH elevada que surgem perante hipotiroidismo congénito primário.
Na criança mais velha, o défice de GH traduz-se por baixa estatura proporcionada e desaceleração progressiva do crescimento, com atraso de idade óssea e por vezes atraso pubertário. Geralmente, no exame físico não são evidenciadas alterações, exceptuando baixa estatura. Alguns casos apresentam aumento da obesidade troncular (“aspecto redondinho”), fácies de “boneca” ou de “querubim” (fronte ampla, hipoplasia da ponte nasal), voz aguda, pele e cabelos finos, característicos do défice congénito de GH. O défice de GH pode associar-se a manifestações clínicas relacionadas com a causa do défice, nomeadamente defeitos da linha média – incisivo central único, fenda palatina, lábio leporino, ou displasia septo-óptica com nistagmo.
Diagnóstico
Uma criança com suspeita clínica de défice de GH deverá ser dirigida a consulta de Endocrinologia Pediátrica. O respectivo diagnóstico implica a verificação de critérios clínicos e auxológicos, bem como confirmação laboratorial. Perante níveis basais de IGF-I diminuídos, o défice hormonal deve ser comprovado por provas de estimulação da produção de GH. Igualmente, deve proceder-se à realização de RM cranioencefálica em todos os casos de défice confirmado para estudo da região hipotálamo-hipofisária e exclusão de patologia do SNC, nomeadamente tumoral.
Tratamento
Em Portugal, o tratamento com GH está sujeito a critérios definidos, sendo os casos submetidos a avaliação por uma Comissão Nacional. A hormona biossintética é administrada diariamente, em injecção subcutânea única, à noite, previsivelmente até ser atingida a idade óssea de 14 anos na rapariga, e de 16 anos no rapaz. Também os critérios de suspensão da terapêutica se encontram definidos.
4. SÍNDROMA DE TURNER
Importância do problema
A síndroma de Turner (ST), ocorrendo em 1/2.500 indivíduos do sexo feminino, deve-se a alteração numérica ou estrutural de um dos cromossomas X. Em cerca de 50% dos casos verifica-se a ausência de um dos cromossomas X (cariótipo 45, X0), em 20-30% ocorrem anomalias estruturais (deleção do braço curto ou do braço longo, cromossomas em anel, isocromossomas); de salientar que em 20-30% dos casos de ST existe mosaicismo (mais frequentemente 45, X0 / 46, XX).
Mais raramente, uma das linhas em mosaicismo pode incluir o cromossoma Y, associando-se o risco acrescido de gonadoblastoma; esta circunstância constitui indicação para gonadectomia profiláctica. De referir que os mosaicos somente são detectados se forem contadas mitoses suficientes ou se forem utilizadas técnicas avançadas de genética molecular. (ver parte referente à Genética).
Manifestações clínicas
O fenótipo da ST é muito amplo em diversidade e gravidade de manifestações. De facto, até 38% dos diagnósticos só são feitos na idade adulta. O cariótipo 45, X tende a associar-se com um fenótipo mais grave do que os mosaicos, mas não existe uma previsível correlação genótipo-fenótipo.
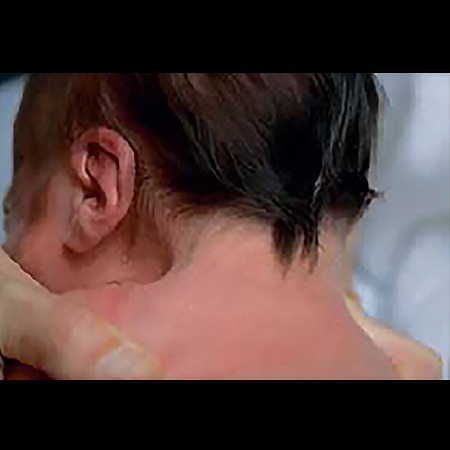
FIGURA 1. Síndroma de Turner. Pescoço curto alado/pterigium colli (NIHDE)
A baixa estatura é um sinal clínico major. Verificando-se em 95-100% dos casos, constitui o achado clínico que mais frequentemente desencadeia a marcha diagnóstica da ST na criança e adolescente. Associada a velocidade de crescimento normal até cerca dos 3 anos, ao longo da infância ocorre desaceleração progressiva do crescimento, o qual persiste além dos 14 anos, fruto da ausência de surto de crescimento pubertário.
Sem tratamento, a estatura final só é atingida na terceira década, e sofre o chamado “efeito Turner” – perda de cerca de 20 cm face ao esperado pela estatura alvo familiar (altura final média de cerca de 143 cm).
Em todas as crianças do sexo feminino com baixa estatura inexplicada dever-se-á considerar a possibilidade de ST, sobretudo perante adolescente com atraso pubertário ou paragem do desenvolvimento pubertário.
A insuficiência gonadal é outra das características nucleares da síndroma, ocorrendo em mais de 90% dos casos. Traduz-se clinicamente por atraso pubertário ou paragem de progressão dos sinais iniciais de puberdade.
No recém-nascido do sexo feminino deve suspeitar-se de ST perante edema linfático das mãos e pés e pele redundante na região posterior do pescoço ou pescoço alado (pterigium colli) (Figura 1).
O Quadro 6 sintetiza a frequência dos múltiplos achados clínicos associados a ST. Destacam-se as alterações cardiovasculares. Os defeitos cardíacos ocorrem em cerca de 1/3 dos casos e atingem mais frequentemente o coração esquerdo: – válvula aórtica bicúspide; – coarctação da aorta; – prolapso da mitral; – mesocárdia; e – aneurisma dissecante da aorta.
Tratamento
O tratamento tem como objectivos:
- aproveitar o potencial de crescimento através da terapêutica com GH biossintética (desde o diagnóstico e na ausência de critérios de exclusão), e também,
- a substituição hormonal necessária à indução de maturação pubertária, replicando o padrão fisiológico (estrogénios a iniciar em idade pubertária, dose inicialmente baixa com aumento progressivo, associando-se posteriormente um progestagénio).
QUADRO 6 – Manifestações clínicas da síndroma de Turner
* Deformidade de Madelung: deformação do punho traduzida por angulações radial e palmar da extremidade distal do rádio | |||||||||||||||||||||
|
| ||||||||||||||||||||
5. CRIANÇAS PEQUENAS PARA A IDADE GESTACIONAL
No âmbito da endocrinologia pediátrica, considera-se por pequeno para a idade gestacional (PIG) todo o recém-nascido com comprimento e/ou peso inferior a -2 DP para a idade e sexo. Em 90% destas crianças verifica-se um processo de crescimento compensatório, atingindo uma estatura final normal. A recuperação do crescimento ocorre, na maioria dos casos, até aos 2 anos de idade, havendo ainda uma pequena probabilidade de recuperação até aos 4 anos. Em 10 a 15% das crianças PIG, o crescimento compensatório é insuficiente, não sendo atingida uma estatura final concordante com a estatura alvo familiar. Em particular, as crianças que não recuperam estatura (não atingindo o percentil da estatura alvo familiar até aos 2 anos de idade), têm maior probabilidade de vir a ter estatura final baixa (< -2 DP).
A realização de exames complementares em situações de BE numa criança PIG deve ser completa – como aliás em qualquer outro caso – visto que podem coexistir outras formas de morbilidade, nomeadamente ST, baixa estatura familiar, défice de GH ou osteocondrodisplasias.
Em tais circunstâncias, os pacientes beneficiam do tratamento com GH, havendo, no entanto, importante variabilidade interindividual na resposta à terapêutica. Salienta-se que a resposta é tanto mais favorável quanto mais cedo for iniciada (a partir dos 4 anos), e quanto mais anos de terapêutica tiverem antecedido a puberdade. Assim, o encaminhamento para a consulta de endocrinologia pediátrica deve ser atempado.
De acordo com as normas da Comissão Nacional para a Normalização da Hormona do Crescimento, constituem critérios para terapêutica com GH em crianças PIG:
- Peso e/ou comprimento ao nascer <-2 DP para a idade gestacional
- Estatura aos 4 anos de idade <-2,5 DP para o sexo e idade
- Diferença entre o DP da estatura da criança e o DP da estatura alvo familiar > 1
- Variação do Z–score da estatura < percentil 50 no último ano (tabelas para consulta na área de especialidade)
– Pré-púbere (volume testicular < 4 mL no rapaz e ausência de botão mamário na rapariga)
BIBLIOGRAFIA
Cheetham T, Davies JH. Investigation and management of short stature. Arch Dis Child 2014;
99: 767-771
Comissão Nacional de Normalização da Hormona de Crescimento (CNNHC). Avaliação de Crianças e Adolescentes com Baixa Estatura. Lisboa Ministério da Saúde 2004
GH Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab 2000; 85: 3990-3993
Hindmarsh PC. Current Indications for Growth Hormone Therapy. London: Karger, 2010
Kliegman RM, Stanton BF, StGeme JW, Schor NF (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2015
Korzeniewski SJ, Allred EN, Joseph RM, et al. Neurodevelopment at age 10 years of children born <28 weeks with fetal growth restriction. Pediatrics 2017;140(5). pii: e20170697. doi: 10.1542/peds.2017-0697. Epub 2017 Oct 13
Mahoney CP. Evaluating the child with short stature. Pediatr Clin North Am 1997; 34: 425-849, 1987
Mercedes O, Winhoven TMA, Onyango WA. Worldwide practices in child growth monitoring. J Pediatr 2004; 1444: 461- 465
Mitchell H, Hindmarsh PC. Assessment and management of short stature. Curr Pediatr 2000: 9: 237-41
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Palminha JM, Carrilho E. Orientação Diagnóstica em Pediatria. Lisboa: Lidel, 2003
Pinsker JE. Turner syndrome: Updating the paradigm of clinical care. J Clin Endocrinol Metab 2012; 97: E994–E1003
Raine JE, Donaldson MDC, Gregory JW, Savage MO, Hintz RL. Short Stature. Practical Endocrinology and Diabetes in Children 2006; 3: 42-64
Rogol AD, Hayden GF. Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J Pediatr 2014; 164 (5 Suppl): S1-S14
Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA (eds). Rudolph’s Pediatrics. New York: McGraw-Hill Medical, 2011
Van der Steen M, Hokken-Koelega AC. Growth and metabolism in children born small for gestational age. Endocrinol Metab Clin North Am 2016; 45: 283-294
Wright CM. The use and interpretation of growth charts. Curr Pediatr 2002; 12: 279-282