Definição
As síndromas neurocutâneas (anteriormente designadas facomatoses – termo derivado de phakos <> lesões na pele, e de oma <> tumorações extracutâneas) são definidas como doenças hereditárias heterogéneas que se caracterizam por anomalias nas estruturas de origem ectodérmica, manifestações cutâneas com tendência tumoral, e alterações do sistema nervoso; outros órgãos como o olho, rim e coração, pulmão e esqueleto são também afectados.
Por estar comprometida a diferenciação e o crescimento celulares (designadamente a diferenciação da ectoderme primitiva), a organogénese está igualmente perturbada, com consequente formação de tumores, geralmente benignos.
As situações que cabem no âmbito da definição são a neurofibromatose, a esclerose tuberosa, a síndroma de Sturge-Weber, a incontinentia pigmenti, a ataxia telangiectasia, a doença de von Hippel-Lindau, a síndroma PHACE, a hipomelanose de Ito e a síndroma de nevus linear.
Formas clínicas
Nesta alínea são abordadas as formas mais frequentes de síndromas neurocutâneas.
1. Neurofibromatose tipo 1 (NF1), ou doença de Recklinghausen, é a síndroma neurocutânea mais frequente, ocorrendo aproximadamente na proporção de 1/3.000 nascimentos. É devida a uma mutação no gene NF1 localizado no cromossoma 17q11.2 o qual codifica uma proteína citoplásmica activadora – a neurofibromina – que actua como supressor ou regulador do crescimento celular e tumoral.
Afecta todos os sexos e raças, e a hereditariedade é autossómica dominante, com penetrância de 98%. A clínica pode ter uma expressão muito variável, sendo o diagnóstico afirmado na presença de determinados critérios (dois ou mais) dos referidos no Quadro 1. Salienta-se que o diagnóstico pode demorar a fazer-se até aos 4-5 anos, quando a doença adquire maior expressividade.
QUADRO 1 – Diagnóstico de neurofibromatose
| Critérios de diagnóstico de neurofibromatose (presença de dois ou mais) |
|
As manchas de tipo “café com leite”, geralmente presentes desde o nascimento, tendem a aumentar em número e tamanho com a idade, ao mesmo tempo que vão surgindo os neurofibromas na pré-puberdade (Figura 1). Associam-se frequentemente ao glioma das vias ópticas (Figura 2) e a estenose do aqueduto de Sylvius. De referir, contudo, que nem todos os indivíduos com tais manchas padecem de neurofibromatose.
Os hamartomas da íris ou nódulos de Lisch raramente se encontram nos primeiros anos de vida, surgindo durante a adolescência. Podem ser identificados através do exame com lâmpadas de fenda.
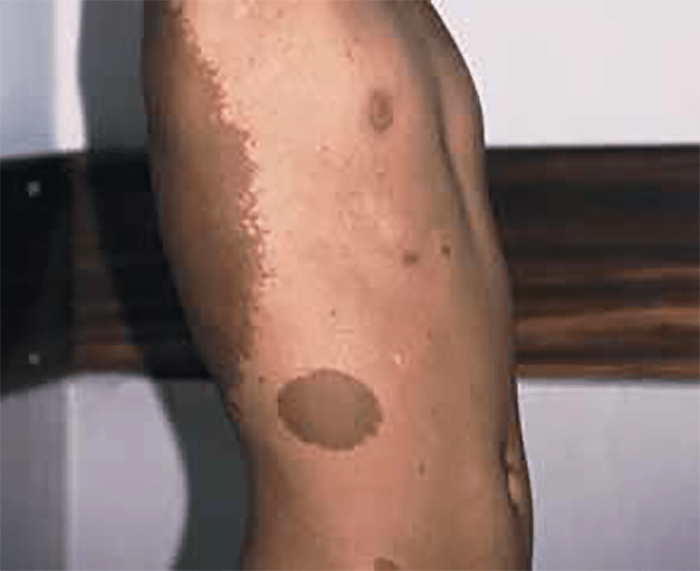
FIGURA 1. Neurofibromatose tipo I. Manchas tipo “café com leite”
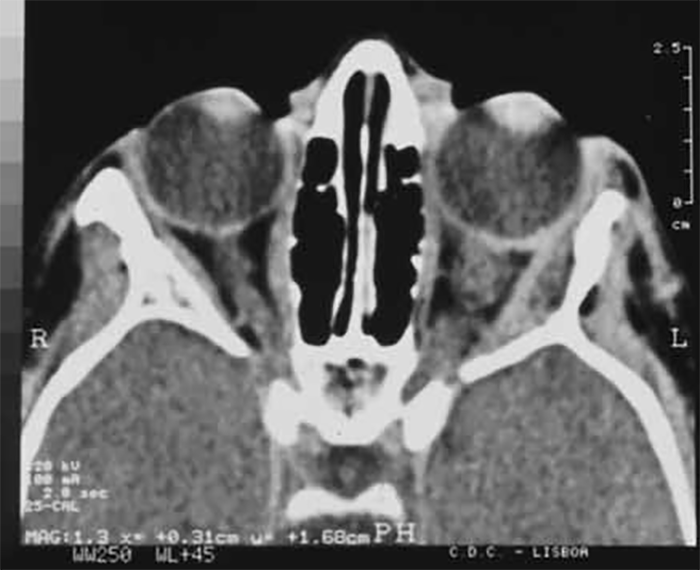
FIGURA 2. Glioma da via óptica (TAC-CE)
A NF1 constitui a forma clínica que mais frequentemente se associa a tumores do sistema nervoso central (SNC), periférico e doutros órgãos, sendo de natureza histológica e de incidência muito variáveis.
Os gliomas da via óptica, os mais frequentes (cerca de 15%), estão presentes desde o nascimento na maioria dos casos (Figura 2). O diagnóstico deve ser feito nos primeiros anos de vida; o exame imagiológico através da ressonância magnética (RM) é o ideal para avaliação das lesões tumorais.
Os tumores do sistema nervoso periférico são neurofibromas e schwannomas, localizados na maioria das vezes na zona de exteriorização das fibras sensitivas no canal raquidiano.
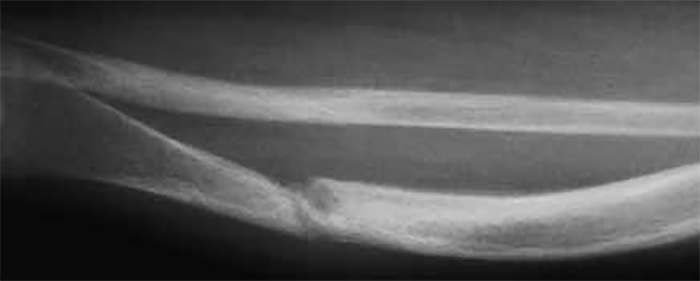
FIGURA 3. Neurofibromatose tipo I: imagem radiológica de pseudartrose.
As alterações ósseas são também frequentes, destacando-se a escoliose com ou sem cifose, a pseudartrose, (Figura 3) a displasia facial (esfenoidal) e, menos frequentemente, a hemi-hipertrofia facial ou generalizada.
As dificuldades escolares são muitas vezes a primeira preocupação dos pais. O défice cognitivo nestes doentes não é habitualmente acentuado, mas estima-se que o quociente de inteligência (QI) se situe entre 15 a 20 pontos abaixo do dos irmãos não afectados. A epilepsia é habitualmente uma complicação menor da NF1 e geralmente de fácil controlo. Outras complicações descritas são a puberdade precoce, além de perturbações endócrinas e a agenesia do corpo caloso.
Neste tipo I de neurofibromatose, advoga-se o tratamento conservador. Quando existem tumores invasivos de crescimento rápido pode tentar-se o tratamento cirúrgico, radioterapia ou quimioterapia, sendo que se gera controvérsia nalgumas modalidades tais como no caso do glioma do nervo óptico.
Na maioria dos casos de NF tipo I a evolução é lentamente progressiva, o que permite sobrevivência significativa. Como se pode depreender, está indicado o seguimento multidisciplinar dada a complexidade da etiopatogénese.
2. Neurofibromatose tipo II (NF2), com uma incidência de 1/50.000 (10% das neurofibromatoses), e manifestando-se após a segunda década de vida, comporta também o modo de transmissão autossómica dominante; podem ocorrer casos esporádicos. O seu gene (NF2), localizado no cromossoma 22q12.2 em 70% dos casos, determina a produção duma proteína anómala chamada merlina. As manchas “café com leite”, clássicas da NF1, podem ou não estar presentes; quando presentes, em menor número relativamente à NF1.
As manifestações iniciais traduzem-se dum modo geral por hipoacusia uni ou bilateral; daí o facto de esta doença ser inicialmente conhecida por neurofibromatose acústica, traduzindo a presença de neurinomas (schwannomas) do acústico unilaterais ou bilaterais. Neste tipo de NF pode verificar-se igualmente o desenvolvimento doutros tumores intracranianos, tais como meningiomas, astrocitomas e schwannomas espinhais.
O diagnóstico obriga à presença dos seguintes critérios: massas bilaterais do VIIIº nervo craniano e história familiar de NF2 com massa do VIIIº nervo craniano unilateral, ou 2 dos seguintes: neurofibroma, meningioma, glioma, schwannoma e cataratas subcapsulares posteriores.
Na maior parte das situações de NF tipo II, opta-se por tratamento conservador, estando indicada a radioterapia perante sinais de malignização. Recentemente nalguns centros tem-se administrado erlotinib para tratamento do schwannoma vestibular com bons resultados.
O prognóstico é muito reservado, em especial pela associação com tumores evolutivos do SNC que recidivam e se multiplicam rapidamente.
3. Incontinentia pigmenti, ou síndroma de Bloch – Sulzberger, afecção dominante ligada ao cromossoma X, atinge sobretudo o sexo feminino (morte in utero dos indivíduos do sexo masculino). Trata-se dum alteração hereditária da mesoderme devida a mutação no gene IKBKG, também denominado NEMO (Xq28.34). O fenótipo resulta de mosaicismo funcional causado por inactivação aleatória do referido gene dominante no cromossoma X, que é letal no sexo masculino.
A afecção, fundamentalmente de expressão dermatológica, caracteriza-se por diminuição ou ausência de melanina nas células basais da epiderme, com incremento da mesma na derme. As lesões da pele passam habitualmente por quatro estádios, desde uma fase inflamatória a outra exclusivamente pigmentada. Em 90% dos doentes na primeira semana de vida (50% dos casos na data do nascimento) surgem lesões eritematosas, vesículas, máculas, pápulas e bolhas, com uma distribuição linear, proximal e predomínio nas superfícies flexoras, acompanhadas de eosinofilia marcada. (Figura 4)
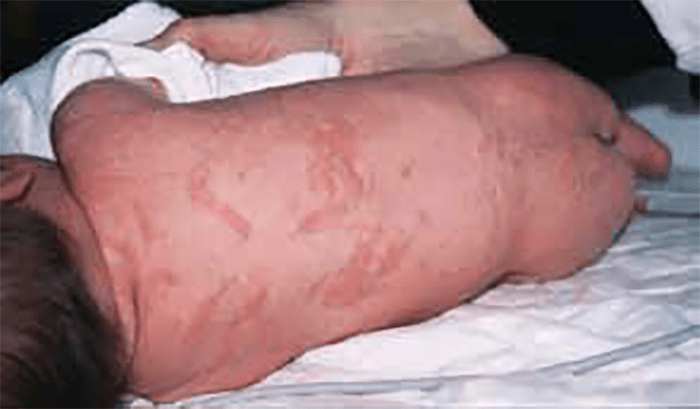
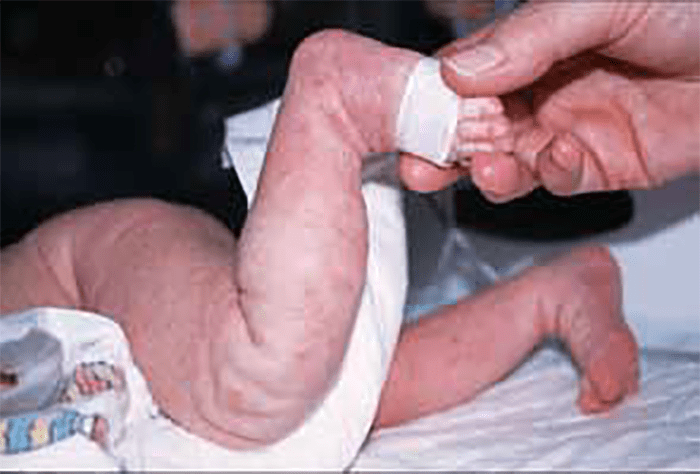
FIGURA 4. Caso de incontinentia pigmenti ou doença de Bloch-Sulzberger. Distribuição linear das lesões cutâneas. (NIHDE)
Mais tarde, estas lesões tornam-se pustulares, queratosas, desenvolvendo-se a pigmentação, usualmente simétrica, de forma espiralada e cor acinzentada ou de chocolate, desaparecendo apenas na segunda ou terceira décadas de vida. Na idade adulta, a presença de máculas hipomelânicas constitui a única manifestação. Quando a doença evidencia esta evolução típica não há necessidade de exame histológico da pele para o diagnóstico.
As manifestações neurológicas, presentes em 30 a 50% dos casos, como epilepsia, atraso mental, paraparésia espástica, microcefalia ou ataxia, constituem as manifestações extradermatológicas mais importantes, na medida em que condicionam o prognóstico. Outras manifestações frequentes são as oculares (em 1/3 dos doentes: retina displásica, pseudoglioma e estrabismo), dentárias (erupção tardia ou dentes cónicos) e ortopédicas (luxação da anca e hemivértebras).
A RM permite demonstrar alterações da substância branca subcortical ou estruturais.
Não existe tratamento específico. De salientar a importância do conselho genético e do seguimento multidisciplinar para detecção de complicações.
4. Síndroma de Sturge-Weber, ou angiomatose encefalotrigeminal, rara, ocorre esporadicamente com uma frequência de 1/50.000. Estão descritos casos de transmissão autossómica recessiva e transmissão dominante. Atinge igualmente os dois sexos e caracteriza-se por anomalias vasculares, habitualmente num processo multissistémico que envolve a pele, SNC, olhos e outros órgãos.
A sua forma completa associa sinais e sintomas relacionados com o angioma leptomeníngeo, o angioma cutâneo e o angioma ocular. (Figura 5)
O angioma cutâneo é um angioma cutaneomucoso facial, cor de vinho do Porto, presente desde o nascimento, que tende a ser unilateral e a envolver a metade superior da face e pálpebra (nevus flammeus), limitado ao território de um ou vários ramos do trigémio. O angioma ocular (da coróide) é ipsilateral, presente em 30% dos casos e pode estar associado a glaucoma.
Da patologia associada, destaca-se a epilepsia em 75 a 90% dos casos, com início no primeiro ano de vida em quase metade destes doentes; a gravidade está muitas vezes relacionada com a localização e extensão da lesão cerebral.
A hemiparésia, (ou hemiplegia), está presente em 30 a 45% dos casos antes dos dois anos e é quase sempre colateral à lesão angiomatosa cerebral.
A insuficiência intelectual afecta > 70% dos doentes e a sua gravidade relaciona-se com a precocidade e gravidade da epilepsia.
O contributo da imagiologia para o esclarecimento do quadro clínico desta situação é importante.

FIGURA 5. Síndroma de Sturge-Weber: angioma cutaneomucoso da hemiface direita e fronte. (NIHDE)
A radiografia simples do crânio pode evidenciar calcificações cranianas a partir da segunda década de vida.
A TAC-CE demonstra precocemente a localização e extensão das calcificações, assim como a hemiatrofia cerebral e a maior captação do contraste na zona angiomatosa e no plexo coroideu homolateral.
A RM com gadolínio permite a visualização em toda a sua extensão da angiomatose meníngea de forma precoce e ainda em fase assintomática.
A angio-RM permite detectar angiomas venosos e lesões trombóticas.
Mediante a PET/tomografia de alta resolução/com emissão de positrões, pode evidenciar-se o hipometabolismo cortical da glucose nas áreas afectadas e estabelecer uma correlação entre a extensão da lesão e o prognóstico.
O prognóstico da síndroma de Sturge-Weber é muito variável e dependente, sobretudo, do controlo das crises, bem como dos défices motor e cognitivo. É muito frequente a evolução para epilepsia refractária à terapêutica médica, razão pela qual a cirurgia da epilepsia deve ser encarada muito precocemente em tais situações.
Com o tratamento estético com laser para o angioma facial têm sido obtidas melhoras parciais. Nos casos de descolamento da retina está indicada a fotocoagulação. O surgimento do glaucoma implica a intervenção do oftalmologista. Tal como nas situações descritas anteriormente, é fundamental a colaboração multidisciplinar.
5. Ataxia telangiectasia, afectando cerca de 1/40.000 nados vivos, pelas suas características clínicas ocupa um lugar importante dentro das doenças degenerativas.
A transmissão é autossómica recessiva, com uma alta incidência de novos casos, por mutações do respectivo gene ATM (11q23.3); de tal resulta uma proteína truncada não funcionante com efeitos diversos, tais como hipersensibilidade a radiações ionizantes, atingimento do processo de reparação do ADN, inibição da sua síntese, incremento de rupturas cromossómicas com consequentes anomalias imunológicas, e incremento da apoptose.
Cursa com ataxia cerebelosa, coreoatetose, telangiectasias oculocutâneas, imunodeficiência, hipersensibilidade às radiações e elevada incidência de neoplasias, como leucemias e linfomas. A ataxia cerebelosa é progressiva, com um início precoce e presente em todos os doentes, (cerca dos 2 anos) enquanto a coreoatetose pode surgir em menos de metade dos mesmos. As telangiectasias oculocutâneas, evidenciando-se geralmente entre os 4-6 anos, afectam de uma forma simétrica a conjuntiva, formando uma rede de finas telangiectasias.
Movimentos oculares anómalos (apraxia óculo-motora), presentes em todos os doentes, podem preceder as telangiectasias. Posteriormente, aparecem as telangiectasias cutâneas (em 40% dos casos), sempre simétricas, na base do nariz, nos pavilhões auriculares ou nas mãos.
O doseamento da alfa-fetoproteína, (elevada), do antigénio carcinoembrionário e das imunoglobulinas (diminuição de Ig A secretória, Ig G2, IgG4 e IgE), são importantes marcadores diagnósticos, associados ao estudo cromossómico e à evolução clínica.
A RM-CE em fases avançadas evidencia atrofia cerebelosa.
O prognóstico está sobretudo dependente da deterioração neurológica. Existe degenerescência espinocerebelosa, lesão dos cornos posteriores da medula com perda dos reflexos tendinosos e atrofia espinal medular: na maioria dos doentes há necessidade de cadeira de rodas entre os 10-15 anos.
A imunodeficiência leva a infecções recorrentes, por vezes graves, interferindo também de uma forma importante no prognóstico.
O tratamento, que não é específico, baseia-se essencialmente na administração de imunoglobulina nos casos de infecções recorrentes, na fisioterapia e na terapia ocupacional. Salienta-se que é importante o diagnóstico precoce, a vigilância clínica atendendo à detecção de eventual surgimento de tumores, o conselho genético e o diagnóstico pré-natal com estudos moleculares.
6. Complexo esclerose tuberosa (CET), ou doença de Bourneville-Pringle, tem uma prevalência de 1/6.000 a 1/8.000, sem diferenças de sexo ou raça. Doença hereditária com ampla variabilidade clínica, devida a anomalia congénita do desenvolvimento embrionário, associa basicamente sinais cutâneos e tumores do SNC.
Transmite-se de modo autossómico dominante (penetrância variável), tendo-se demonstrado mutações espontâneas em 60-80% dos casos. Foram identificados 2 loci génicos, TSC1 no cromossoma 9 (9q34.3) e TSC2 no 16 (16p13.3). A incidência de novas mutações é muito elevada.
A patogénese desta anomalia reside na presença de tuberosidades corticais (que deram o nome à doença), nódulos subependimários e tumores de células gigantes, juntamente com anomalias da migração, proliferação e diferenciação neuronais.
Sob o ponto de vista anatomopatológico encontram-se no cérebro tuberosidades corticais formadas por nódulos de tamanho variável, com células gigantes, redução do número de neurónios e aumento dos núcleos astrocíticos, e nódulos subependimários formados por células astrocíticas fusiformes com deposição cálcica fazendo, no seu conjunto, procidência para dentro do ventrículo.
As manifestações clínicas que habitualmente conduzem ao diagnóstico são cutâneas, podendo existir também neurológicas, retinianas, cardíacas e renais.
O atingimento cutâneo é constante. As manchas cutâneas hipopigmentadas (90% dos doentes), poligonais ou em forma de folha ou ponta de lança, podem estar presentes desde o período neonatal ou infância precoce. Podem observar-se à vista desarmada ou com lâmpada de Wood.
O angiofibroma facial ou adenoma sebáceo (em > 70% dos doentes) compreende um conjunto de nódulos rosados no nariz, região malar e região nasogeniana (os chamados nódulos de Pringle), de tamanho variável, entre o da ponta duma agulha e de uma lentilha. Estas lesões podem aparecer já na idade pré-escolar.
Podem coexistir fibromas ungueais, periungueais (tumores de Koenen) e na mucosa oral, falhas no esmalte dentário em forma de fossetas, e lesões de despigmentação tipo serpentina ou madeixas de cabelos brancos.
As manifestações neurológicas – que podem preceder ou surgir em simultâneo com as cutâneas – são a epilepsia (80-90%), o défice cognitivo (60-70%) e as alterações do comportamento como défice de atenção e hiperactividade, autismo, agressividade e psicose.
Os tumores benignos resultantes da proliferação glial são mais frequentes no córtex cerebral, gânglios da base e paredes dos ventrículos. Os nódulos subependimários de maiores dimensões podem condicionar hidrocefalia (ver atrás).
Quanto às manifestações oculares destacam-se os hamartomas retinianos e as manchas hipopigmentadas na íris.
Os rabdomiomas cardíacos (30-70% dos casos) são hamartomas que tendem a ser múltiplos, podendo ser detectados por ecocardiograma fetal e desaparecer espontaneamente nos primeiros anos de vida.
Outras manifestações sistémicas do CET são o angiomiolipoma (75% dos casos) ou quistos renais, a linfangiomatose pulmonar com formação de quistos, pólipos hamartomatosos do recto, lesões ósseas quísticas e alterações endócrinas como puberdade precoce, doenças da tiroideia e gigantismo.
Para o diagnóstico consideram-se as chamadas características major e as minor.
As características major incluem: lesões cutâneas, cerebrais, oculares, e tumores no coração, rins ou pulmões.
As características minor incluem: quistos ósseos, pólipos rectais, alterações do esmalte dentário, anomalias do SNC (alterações da migração celular na substância branca), fibromas gengivais, hamartomas não renais, alterações despigmentares da retina, lesões cutâneas e quistos renais múltiplos.
O diagnóstico definitivo do CET faz-se em função da presença de 2 ou mais critérios major ou 1 major e 1 minor. O diagnóstico provável faz-se, se existir 1 critério major e 1 minor.
Na avaliação diagnóstica destes doentes é fundamental a imagiologia cerebral (TAC ou, de preferência, RM), EEG, ecografia renal, ECG, ecocardiograma, radiografia do tórax, etc..
O tratamento e o prognóstico são variáveis e dependem, não das manifestações cutâneas, mas essencialmente do aparecimento de tumores internos.
A verificação de hipertensão intracraniana relacionável, por ex. com obstrução do buraco de Monro, poderá estabelecer a indicação de intervenção neurocirúrgica urgente.
7. Síndroma PHACE
Esta síndroma agrupa um conjunto de anomalias a que correspondem as letras da sigla PHACE, a saber: anomalias da fossa Posterior, Hemangiomas, anomalias Arteriais, Coarctação da aorta, e outras anomalias – cardíacas e oculares (Eye).
Verifica-se predomínio no sexo feminino. Os hemangionas da via aérea podem originar obstrução. O interferão-alfa tem sido empregue para tratamento dos hemangiomas.
8. Angiomatose cerebelorretiniana (doença de von Hippel-Lindau)
Esta doença transmite-se com carácter autossómico dominante e penetrância variável. A anomalia relaciona-se com o gene VHL (3p25-26), o qual codifica duas proteínas supressoras de tumores.
As manifestações clínicas são marcadas fundamentalmente pela presença de hemangioblastomas do cerebelo e angioblastomas da retina a partir dos 10 anos de idade. A sintomatologia integra sinais agudos de disfunção cerebelosa e policitémia devida à produção de eritropoietina pelo tumor.
As complicações podem surgir a vários níveis: compressão medular por hemorragia, descolamento da retina, laucoma secundário, quistos congénitos do pâncreas e rim, hipernefroma ou feocromocitoma.
O tratamento é cirúrgico e o prognóstico depende da presença e dimensões dos tumores intracranianos ou abdominais. Actualmente nalguns centros especializados têm sido aplicados inibidores da angiogénese nos tumores extraneurais.
AGRADECIMENTOS
Os autores e editor agradecem ao Dr. Raul Silva a cedência das imagens das Figuras 1, 2 e 3.
BIBLIOGRAFIA
Aicardi J (ed). Diseases of the Nervous System in Childhood. London: Mac Keith Press, 2009
Barros FS, Marussi VHR, Amaral LLF, et al. The rare neurocutaneous disorders: update on clinical, molecular, and neuroimaging features. Top Magn Reson Imaging 2018; 27: 433-462. doi: 10.1097/RMR.0000000000000185
Campagnoni AT, et al (eds). Developmental Neuroscience. Basel: Karger, 2008
Campistol J (ed). Neurologia para Pediatras. Madrid: Panamericana, 2011
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. NEJM 2006; 355: 1345-1356
DiMario FJ Jr. Brain abnormalities in tuberous sclerosis complex. J Child Neurol 2004; 19: 650-657
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Korf BR, Bebin EM. Neurocutaneous disorders in children. Pediatr Rev 2017; 38:119-128
Mann JA, Siegel DH. Common genodermatosis: what the pediatrician needs to know. Pediatr Ann 2009; 38: 91 – 99
McLone DG. Pediatric Neurosurgery. Philadelphia: Saunders, 2001
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Osborne JP, Merrifield J, O’Callaghan JK. Tuberous sclerosis – what’s new? Arch Dis Child 2008; 93: 728 – 731
Roach ES (ed). Pediatric Neurology. Philadelphia: Elsevier, 2019
Swaiman KF, Ashwal S, Ferriero DM, Schor NF. Swaiman’s Pediatric Neurology. Principles and Practice. Philadelphia: Elsevier Saunders, 2012
Williams VC, Lucas J, Babcock MA, et al. Neurofibromatosis type I revisited. Pediatrics 2009; 123: 124 – 133