Bases anatomofisiológicas e neurodesenvolvimento
A retina, considerada um prolongamento do SNC, é a camada mais interna das três que constituem a parede de globo ocular.
A coroideia, um dos três constituintes da úvea, é uma estrutura vascular responsável por uma parcela importante do suprimento sanguíneo e nutritivo à retina.
O vítreo, estrutura transparente, propaga a luz submetida a refracção pela córnea e cristalino, transmitindo-a à retina. Posteriormente a retina continua-se pelo nervo óptico.
Quanto a aspectos histológicos, a retina pode dividir-se em duas porções: a retina neurossensorial e a camada de epitélio pigmentado. A primeira é composta por nove camadas onde se dispõem os fotorreceptores e as células relacionadas com a transmissão dos impulsos nervosos e respectivos dendritos e axónios. (Figura 1) A camada de epitélio pigmentado contacta com a coroideia. As duas porções, com origem embriológica diferente, resultam de dois folhetos diferentes de neuroectoderme, formando-se entre os mesmos um espaço subretiniano, virtual, que se une atrás (no rebordo da papila óptica) e à frente (ora serrata).
O desenvolvimento da retina inicia-se na quarta semana de gestação, com a invaginação da vesícula óptica, uma estrutura derivada do diencéfalo. Durante o quarto mês inicia-se a vascularização retiniana definitiva que substitui o sistema vascular primitivo. A vascularização retiniana definitiva, a partir da papila óptica, atinge a periferia temporal no nono mês de gestação. Numa gestação de termo, a retina já se encontra bem diferenciada, havendo completa maturação celular com excepção da mácula, centrada pela fóvea. Esta zona da retina, responsável pela visão central de alta resolução, continua a desenvolver-se atingindo a configuração adulta por volta dos 4 anos. O desenvolvimento da fóvea coincide com o crescimento dendrítico cortical e formação de sinapses.
As funções da retina são transformar a imagem óptica em sinais eléctricos, (o que é executado pelos fotorreceptores), e processar as características do mundo visual, transmitindo os sinais captados pelos fotorreceptores até ao córtex visual através do nervo óptico.
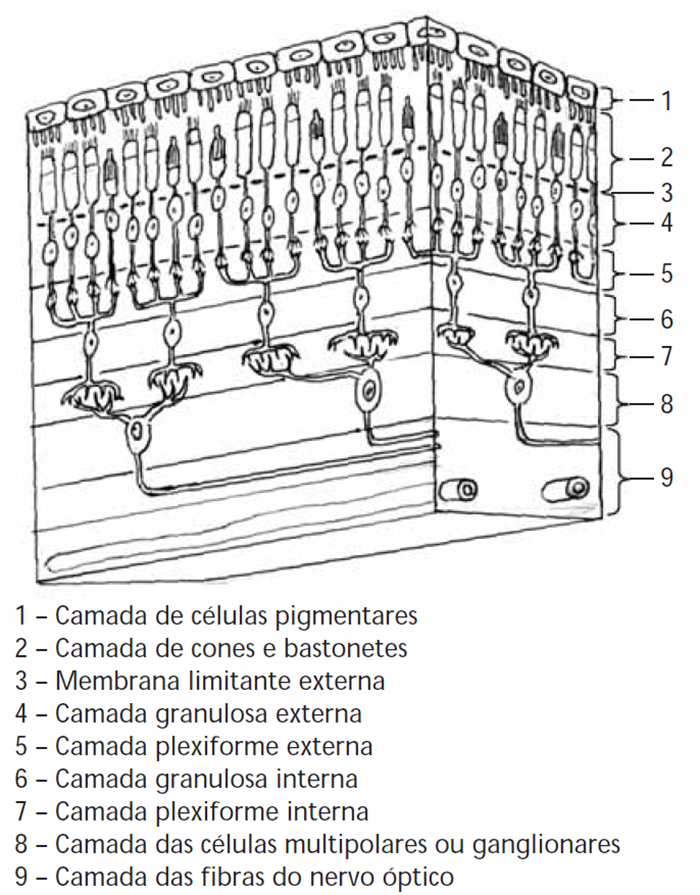
FIGURA 1. Camadas da retina: representação esquemática
Manifestações clínicas
As doenças da retina habitualmente manifestam-se por perturbações da visão, baixa da acuidade ou alteração da qualidade visual. Se na criança que já se sabe exprimir o aparecimento dos sintomas pode ser verbalizado, na criança mais pequena são os sinais ou as complicações de determinada situação patológica que podem sugerir o diagnóstico de doença retiniana. Eis algumas manifestações a realçar:
- Diminuição da acuidade visual – Quando a criança não sabe exprimir a perda de acuidade ou não valoriza o sintoma por ser muito precoce, o comportamento pode ser sugestivo: desinteresse por objectos distantes, não reconhecimento de faces conhecidas a partir de certa distância, sinal oculodigital de Franceschetti (procura de estímulo visual pela compressão dos globos oculares), dificuldade ou atraso na
- Alteração da qualidade visual – Traduz-se por sinais variados que podem apontar para problemas clínicos específicos:
- Discromatopsia – Alteração da visão das cores, por anomalia dos cones ou neuropatia óptica. Pode ser congénita e não evolutiva como o daltonismo, ou adquirida e frequentemente evolutiva, devendo-se a patologia coriorretiniana ou iatrogenia medicamentosa;
- Hemeralopia ou cegueira diurna – Diminuição da acuidade visual em condições de luz Tal alteração sugere doença degenerativa da retina (cones), podendo também dever-se a opacidade da córnea ou cristalino;
- Nictalopia ou cegueira nocturna – Dificuldade visual em condições de baixa luminosidade. O medo extremo do escuro e a dificuldade na orientação em ambientes escurecidos podem ser os sinais reveladores. Este problema surge nas degenerescências pigmentares da retina;
- Fotopsia – Sensações luminosas (por ~”faíscas”) produzidas por estímulos não ópticos nem luminosos, como a excitação mecânica induzida pela pressão digital no globo ocular ou a tracção do vítreo;
- Miodesopsia – Percepção de manchas escuras (~”moscas volantes”) no campo visual que parecem flutuar com os movimentos dos Podem dever-se à presença de exsudado inflamatório ou a sangue no vítreo (hemovítreo), assim como ao descolamento retiniano, situações que projectam sombras sobre a retina;
- Metamorfopsia – Percepção de imagem distorcida, aumentada ou diminuída. Deve-se a doença macular, associando-se habitualmente a baixa da acuidade visual.
- Alterações campimétricas – Perda de campo visual que pode assumir várias formas, tais como: ser localizada a uma área (escotoma), generalizada, etc., consoante a quantidade e localização de fibras nervosas afectadas.
- Leucocória – Trata-se de “pupila de cor branca” a qual pode ter várias causas; relaciona-se com opacidade da córnea, do cristalino (catarata), com patologia do segmento posterior do globo ocular (retinoblastoma, descolamento da retina de causa inflamatória, infecciosa, degenerativa, traumática, ), patologia vítreo-retiniana ou defeito congénito.
- Estrabismo – O estrabismo pode ser a única manifestação de doença retiniana; assim, qualquer criança estrábica deve ser sempre avaliada e sujeita a A baixa de visão traduz-se em dificuldade na fixação e consequente desvio do olho.
- Nistagmo – A sucessão de movimentos rítmicos, involuntários e conjugados dos globos oculares, com alternância de oscilações lentas e rápidas pode ser uni ou bilateral. Define-se, convencionalmente, pelo sentido da oscilação rápida e pela sua direcção: horizontal, vertical, rotatório, multidireccional ou misto. Constitui sinal de lesões do aparelho vestibular ou das vias nervosas centrais ou periféricas; pode ser provocado por certas posições. Quando de origem retiniana ocorre por incapacidade de fixação associada a patologia macular.
Principais doenças retinianas
Em termos semiológicos as doenças retinianas podem dividir-se em dois grupos: as doenças maculares e as doenças da periferia retiniana. Recorda-se que a mácula é a região da retina que contribui primordialmente para a visão central, fotópica e de alta resolução.
As doenças maculares caracterizam-se por diminuição da acuidade visual, metamorfopsia, fotofobia, hemeralopia e alterações campimétricas centrais (escotoma central). A periferia retiniana é responsável pela visão periférica, escotópica e de orientação espacial, pelo que as afecções periféricas se associam à perturbação da referida visão.
A avaliação objectiva das doenças retinianas compreende a medição da acuidade visual (acuidade de detecção medida com pequenos objectos, acuidade de padrões pelo olhar preferencial ou pela resolução de linhas de orientação, acuidade de reconhecimento com testes de optótipos e acuidade de leitura), determinação da sensibilidade ao contraste (capacidade de ver detalhes em níveis baixos de contraste), registo dos campos visuais e avaliação da visão cromática. Completa-se pela visualização directa da retina (oftalmoscopia directa e indirecta), habitualmente com a pupila dilatada farmacologicamente. Frequentemente é necessário recorrer a outros exames complementares como a retinografia, a angiografia fluoresceínica, a ecografia ocular, a tomografia de coerência óptica e a electrofisiologia (electrorretinograma, electroculograma e potenciais evocados visuais).
São referidos a seguir problemas clínicos de etiopatogénese diversa associados a doença retiniana, chamando-se especial atenção para a retinopatia da prematuridade e para o retinoblastoma.
Anomalias congénitas
As anomalias congénitas da retina e nervo óptico podem implicar um compromisso funcional variável, salientando-se que algumas delas poderão indiciar a existência de defeitos com outra localização. Surgem com uma incidência ~1,6/1.000 em RN.
São exemplos a displasia vítreo-retiniana, a persistência do vítreo primário hiperplásico, anomalias vasculares e o coloboma (qualquer anomalia congénita do desenvolvimento que poderá surgir em qualquer das seguintes estruturas: pálpebras, íris, cristalino, coroideia, retina ou nervo óptico).
O coloboma é um dos constituintes da associação CHARGE, razão pela qual a sua verificação obriga à detecção doutros defeitos (síndromas malformativas, cromossomopatias, etc.).
Albinismo
O albinismo corresponde a um grupo heterogéneo de doenças, quer sob o ponto de vista genético, quer clínico; caracteriza-se por hipopigmentação da pele, cabelo e olhos, explicável por deficiência da produção de melanina. São descritas duas formas de albinismo: oculocutâneo e ocular. As síndromas de Hermansky-Pudlak e Chediak-Higashi associam-se a albinismo oculocutâneo.
Doenças hereditárias do metabolismo
Existem mais de 400 doenças hereditárias com envolvimento significativo da retina, mácula ou coróide, com quadros clínicos diversos de degenerescência retiniana, conforme as áreas inicialmente atingidas e progressão verificada. A patologia metabólica pode originar alterações retinianas com aspectos particulares, nomeadamente as degenerativas (retinopatia pigmentar, atrofia girata, alteração do epitélio pigmentar e alteração da mácula em mancha cor de cereja), bem como quadros de hemorragia retiniana e neuropatia óptica. (Quadro 1)
QUADRO 1 – Degenerescência retiniana e doenças hereditárias do metabolismo
| * LCHAD – Hidroxiacil-CoA desidrogenase dos ácidos gordos de cadeia longa |
| Doença retiniana associada a outras doenças |
|
| Doença retiniana isolada |
|
Nas alterações degenerativas da mácula, o aspecto em mancha cor de cereja é de uma forma geral relacionado com etiologia metabólica, sendo fundamental o diagnóstico diferencial das doenças de sobrecarga lisossomal. Tal padrão do fundo ocular é devido à acumulação de lípidos complexos nas células ganglionares da retina, originando uma cor esbranquiçada rodeando a zona da fóvea. (Figura 2 e Quadro 2)
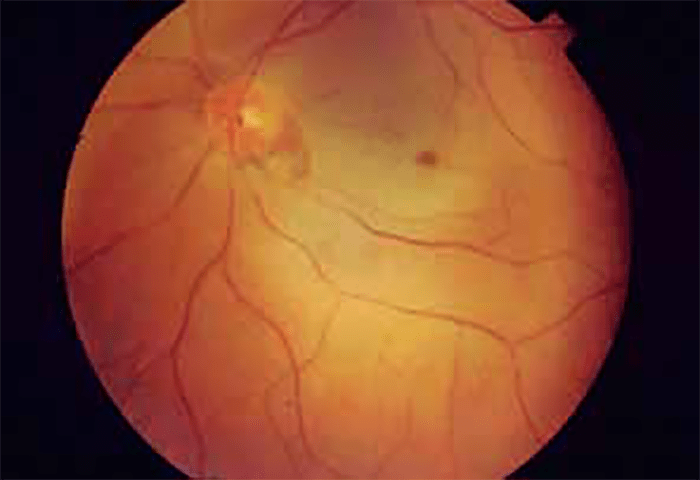
FIGURA 2. Fundoscopia: mancha cor de cereja
QUADRO 2 – Degenerescência macular em “mancha cor de cereja”
| Doença | “Mancha cor de cereja” |
| Sialidoses tipo I | Constante |
| Sialidoses tipo II | Constante |
| Galactossialidose | Frequente |
| Gangliosidose GM2 | |
| • Doença de Tay-Sachs (infantil) | Constante |
| • Doença de Sandhoff (infantil) | Constante |
| Gangliosidose GM1 (infantil) | Frequente |
| Doença de Niemann-Pick tipo A | Frequente |
| Doença de Gaucher tipo 2 | Ocasional |
Diabetes mellitus
No que se refere a diabetes mellitus (tipos 1 e 2) cabe referir que as manifestações oculares mais frequentes ocorrem na retina e no cristalino. A retinopatia diabética raramente surge antes de 3-5 anos de doença, e antes da puberdade. O tempo de evolução e o grau de descompensação metabólica da doença são factores relevantes no desenvolvimento e na gravidade das complicações oculares. Surge microangiopatia progressiva que leva à lesão e oclusão dos pequenos vasos retinianos. Em fases inicias da retinopatia (chamada de fundo), à fundoscopia observam-se microaneurismas (dilatação capilar), tortuosidade vascular, hemorragias e edema. Pode haver défice da aquidade visual se o edema atingir a mácula.
Com o tempo, a oclusão microvascular progressiva leva à isquémia e à formação de neovasos (retinopatia proliferativa), com edema e hemorragia mais acentuados, (hemorragias retinianas e do vítreo), maculopatia e risco mais elevado de descolamento retiniano. A retinopatia é lentamente progressiva, sabendo-se que, após 10 anos de doença, 70 a 90% dos diabéticos do tipo 1 evidenciam graus diversos de retinopatia, sendo tanto mais grave quanto pior o controlo metabólico.
O compromisso associado do cristalino, evidenciado pela sua opacificação (catarata) depende também muito da qualidade e estabilidade da compensação metabólica. A presença de catarata diminui a acuidade visual.
A intervenção cirúrgica está indicada no tratamento da catarata, do hemovítreo ou do descolamento retiniano. O método de tratamento com raios laser é utilizado como profilaxia destas últimas complicações e no tratamento de certas formas de maculopatia.
O acompanhamento oftalmológico da criança e do adolescente diabético deve ser ajustado à idade e à gravidade da diabetes. Em geral, deve fazer-se a avaliação uma vez feito o diagnóstico, com a finalidade de esclarecimento da criança/família e exclusão de outra patologia. A detecção e vigilância da retinopatia deve iniciar-se 3 anos após o início do período da puberdade, quando a evolução metabólica tenha sido favorável, ou antes, pelos 9 anos de idade, no caso de não ser conseguido tal objectivo. Posteriormente, a vigilância oftalmológica deve ser, pelo menos, semestral.
Doenças hematológicas
A retinopatia verificada nos casos de doença de células falciformes relaciona-se com a hiperviscosidade sanguínea e consequente hipóxia, salientando-se que as manifestações oculares são mais frequentes quando há associação a outra hemoglobinopatia.
As lesões encontram-se na periferia, podendo configurar uma forma não proliferativa, de fundo, caracterizada por envolvimento arteriolar com embainhamento, tortuosidade e hemorragias (manchas-salmão, intrarretinianas, e aglomerados pigmentares cicatriciais), ou evoluir para uma forma proliferativa quando a isquémia é mantida. A retinopatia drepanocítica proliferativa (rara em idade pediátrica) deve-se à oclusão arteriolar e neovascularização; define-se pela presença de tufos vasculares de aspecto típico e pode levar a complicações irreversíveis.
A aplicação do método de laser pode estar indicada como medida preventiva.
A vigilância oftalmológica deve iniciar-se por volta dos 10 anos de idade e repetir-se semestralmente.
Inflamação e infecção
A inflamação da retina habitualmente não ocorre de forma isolada, associando-se, pelo menos, ao compromisso da coroideia. Daí que os processos inflamatórios destas duas estruturas sejam habitualmente considerados em simultâneo, mesmo quando um processo é predominantemente retiniano.
O termo uveíte significa inflamação da úvea (estrutura composta pela íris, corpo ciliar e coroideia). Se todas as referidas estruturas estiverem atingidas, diz-se que há panuveíte.
Segundo o critério topográfico, são considerados três tipos de uveíte:
- anterior, compreendendo a irite e a iridociclite;
- intermédia, compreendendo a ciclite e a chamada pars planite;
- posterior, compreendendo a coroidite (ou a coroidorretinite quando se verifica compromisso retiniano); ou seja, a uveíte posterior atinge predominantemente o segmento posterior do globo ocu
Quanto à etiologia, podem ser considerados quatro grupos: de origem infecciosa, autoimune e idiopática, para além de situações integrando as chamadas “síndromas mascaradas”. Estas últimas compreendem situações clínicas diversas que cursam com uveíte, por ex.: linfoma, retinoblastoma, leucemia, metástases de tumor distante, descolamento, degenerescência retiniana e presença de corpo estranho intra-ocular.
A uveíte intermédia é, em geral, uma situação crónica e idiopática.
Na idade pediátrica, cerca de 40% das uveítes afectam o segmento posterior, sendo a etiologia infecciosa (congénita ou adquirida) a mais importante: por Toxoplasma gondii, Citomegalovirus, Mycobacterim tuberculosis, Candida albicans, Toxocara canis, etc..
Como noutras doenças retinianas, a presença de uveíte posterior pode revelar-se quando a criança é capaz de expressar queixas, nomeadamente de miodesopsia, de alteração da acuidade ou qualidade visual, ou através de sinais que revelam diminuição da acuidade visual. Frequentemente, trata-se de situações que se associam a outras manifestações oculares, inflamatórias ou a anomalias congénitas.
O atraso no diagnóstico devido à não verbalização das queixas e as consequências sobre o sistema visual, ainda em desenvolvimento, podem agravar o prognóstico na criança mais pequena.
Refira-se, no entanto, que por vezes o diagnóstico de uveíte posterior é um achado ocasional no âmbito de observação oftalmológica realizada por motivos diversos.
São descritos a seguir alguns quadros clínicos que tipificam processos de inflamação e infecção de expressão retiniana:
- Toxocarose – Deve-se à presença da larva enquistada na Em geral existe compromisso uniocular. O aspecto mais típico é o de granuloma localizado no pólo posterior do olho ou na periferia; no entanto, o processo inflamatório pode ser mais difuso. Ao cicatrizar, pode originar o descolamento da retina. As manifestações clínicas incluem diminuição de visão, estrabismo ou leucocória, impondo por vezes, o diagnóstico diferencial com retinoblastoma.
- Retinites víricas – Vários agentes víricos (por vírus da imunodeficiência humana/VIH, Herpes simplex, Herpes zoster, Citomegalovirus e vírus da rubéola) podem infectar a retina da criança, desde a fase de vida intra-uterina, podendo as respectivas repercussões ser mais ou menos devastadoras. Para além da infecção da retina, outras estruturas oculares podem estar envolvidas, como a córnea, o cristalino, a íris, o corpo ciliar, o nervo óptico, etc..
O exame fundoscópico permite observar focos activos de coriorretinite (áreas de edema retiniano que se podem associar a fenómenos de vasculite e hemorragias) ou cicatrizes de infecção coroidorretiniana (áreas de hiperpigmentação alternando com áreas menos pigmentadas ou esbranquiçadas) em maior ou menor extensão. Um aspecto frequente nalgumas infecções congénitas (rubéola, sarampo) é o quadro designado classicamente por “retinopatia pigmentada em sal e pimenta” em que se observa a dispersão pigmentar em toda a retina ou parte dela.
A doença ocular em crianças infectadas pelo VIH e Citomegalovirus é muito menos frequente do que nos adultos. - Coroidorretinites bacterianas e fúngicas – Trata-se de situações mais raras, habitualmente ocorrendo associadas a outras manifestações oculares e não A sífilis congénita pode dar origem a retinopatia em sal e pimenta semelhante à da rubéola. O envolvimento retiniano é ainda possível na doença de Lyme, na tuberculose e na doença do arranhão do gato. Em todas estas doenças as manifestações clínicas são muito variáveis, não havendo um padrão típico de apresentação.
A retinopatia surgindo no contexto de septicémia ou bacteriémia é muito rara; em geral deve-se a um êmbolo séptico decorrente de endocardite bacteriana. O exame oftalmológico especializado permite identificar sinais de hemorragia centrada por uma área branca (mancha de Roth), como manifestação do êmbolo formado. As infecções por fungos (por ex. Candida albicans) também podem originar endoftalmite por embolia. - Coroidorretinite por Toxoplasma gondii (Toxoplasmose) – A toxoplasmose (congénita ou adquirida) é a causa mais importante de uveíte posterior em todos os grupos etários; em idade pediátrica é responsável por cerca de 50% dos casos de uveíte posterior (coroidite ou coroidorretinite). Uma vez instalado na célula retiniana, há proliferação do protozoário causando reacção de hipersensibilidade e inflamação nos tecidos e vasos adjacentes, vítreo e coroideia, sendo de realçar que poderá surgir reactivação em zona adjacente a cicatriz coroidorretiniana antiga. As manifestações de toxoplasmose congénita variam muito, entre cicatriz retiniana periférica e coroidorretinite activa.
- Uveítes não infecciosas – Estas afecções são muito raras em idade pediátrica salientando-se, a este propósito, que alguns casos de uveíte posterior podem associar-se a sarcoidose, a nefrite tubulointersticial (TINU) e a doença de Behçet.
Distrofias coroidorretinianas
Abrangem um conjunto de situações de natureza genética relativamente às quais cada vez são conhecidos mais genes; geralmente só se manifestam no adolescente ou adulto. Para o estabelecimento do diagnóstico e prognóstico destas doenças, a electrofisiologia ocular é fundamental. Outros meios auxiliares de diagnóstico importantes são o estudo da visão cromática e a angiografia fluoresceínica. São salientadas, entre outras, as seguintes distrofias:
- Retinopatia pigmentada (retinose pigmentar) – É caracterizada por disfunção progressiva dos fotorreceptores e atrofia de várias camadas da retina, verificando-se perda progressiva de bastonetes e, posteriormente, também de cones. Manifesta-se essencialmente por nictalopia e diminuição progressiva dos campos visuais. Por fundoscopia são identificadas alterações pigmentares retinianas típicas (espiculadas), atenuação vascular e palidez óptica. Na maioria dos casos, o modo de transmissão é autossómico recessivo, tendo sido comprovada hereditariedade ligada ao cromossoma X em cerca de 15% dos casos.
- Distrofia de cones-bastonetes – Nesta forma os fotorreceptores são afectados de forma generalizada, sendo maior a perda de função do cone do que a do bastonete. A hereditariedade é autossómica (recessiva ou dominante), ou ligada ao cromossoma X. A maioria revela-se entre o final da segunda década de vida e o início da idade adulta. Manifesta-se essencialmente por perda de visão central, discromatopsia e fotofobia, instalando-se progressivamente nictalopia e perda de visão periférica. Por fundoscopia observam-se alterações semelhantes às da retinopatia pigmentada.
- Amaurose congénita de Leber – Trata-se duma distrofia cone-bastonete identificável desde a data do nascimento ou nos primeiros meses de vida por défice de visão e nistagmo. Em geral, a transmissão é autossómica recessiva, podendo também ocorrer a forma autossómica dominante. Constitui a causa mais frequente de deficiência visual infantil de natureza hereditária. As manifestações ulteriores incluem movimentos erráticos dos globos oculares ou nistagmo, deficiente reflexo fotomotor pupilar e o já citado sinal oculodigital de Franceschetti (como resultado da deficiente estimulação visual pela luz, a criança exerce pressão sobre os globos oculares para estimular mecanicamente a retina, conseguindo, assim, obter sensação visual); com o tempo, a pressão repetida pode levar a atrofia da gordura orbitária e afundamento do globo ocular – enoftalmia – e a catarata.
Descolamento da retina
Entende-se por descolamento retiniano a separação entre a retina neurossensorial e o epitélio pigmentado subjacente por mecanismos diversos. Como consequência, a retina pode sofrer atrofia por insuficiência de suprimento sanguíneo e de nutrientes.
As manifestações clínicas essenciais variam consoante a causa do descolamento e a sua extensão: fotopsias, miodesopsias, perda de campo ou acuidade visual, e leucocória. Como em qualquer situação que decorre com perda de acuidade visual, o estrabismo adquirido pode ser um sinal revelador.
Retinopatia da prematuridade
Definição
Entende-se, por retinopatia da prematuridade (RP) a doença vascular retiniana consequente à proliferação fibrovascular anómala numa retina em desenvolvimento, com vascularização incompleta. A prematuridade, pelas repercussões na maturação do globo ocular e das estruturas do sistema nervoso central, pode ter implicações no desenvolvimento da visão. Neste contexto, as principais alterações oftalmológicas com as quais o clínico depara são, a chamada retinopatia da prematuridade na fase aguda, e suas sequelas: os defeitos refractivos e o estrabismo.
Aspectos epidemiológicos e importância do problema
A RP é uma doença cuja incidência e gravidade são inversamente proporcionais ao peso de nascimento e à idade gestacional. De acordo com o CRYO-ROP Group, a frequência é ~ 47% em RN com idade gestacional > 31 semanas e peso entre 1.000 e 1.250 gramas; e ~90% nos casos com < 28 semanas e < 750 gramas. Estudos mais recentes apontam para uma diminuição da incidência de retinopatia (10 a 40%) e redução da probabilidade de ocorrência de retinopatia limiar (3%); no entanto, em absoluto, os casos graves são cada vez mais frequentes, dado o aumento de sobrevivência de RN com peso de nascimento inferior a 750 gramas em virtude dos progressos realizados em terapia intensiva.
Etiopatogénese
A angiogénese dos vasos da retina inicia-se pelas 16 semanas de gestação, prolongando-se até cerca das 40 semanas. Tem a sua origem no pólo posterior do globo ocular, ao nível da papila óptica, evoluindo anteriormente até atingir a ora serrata, o limite anterior da retina. Por outras palavras: a vascularização da retina do feto faz-se da papila óptica para a periferia; a mesma completa-se primeiramente na retina lado nasal pelo facto de esta ser mais curta. Por isso, é mais frequente o compromisso da retina (RP) no lado temporal cujo desenvolvimento se completa pelas 40 semanas (gestação de termo).
Quando a criança nasce após gravidez muito encurtada em tempo (recém-nascido pré-termo), o processo de angiogénese não se encontra completo.
A RP progride em duas fases. Numa primeira fase (hiperóxica), o contacto com meio ambiente extra-uterino, mais rico em oxigénio que o meio intra-uterino, cria vasoconstrição que, se prolongada, se torna irreversível, tendo em conta a elevada susceptibilidade dos vasos retinianos a elevada pressão parcial de oxigénio*.
| *No sangue fetal a saturação em O2 da Hb é ~70%, enquanto no RN de termo respirando ar é ~100% (correspondendo respectivamente a PaO2 de 30 mmHg e de 60-100 mmHg). |
Numa segunda fase (isquémica) – que surge à medida que o RN pré-termo cresce (entre as 30 e as 34 semanas de idade gestacional) – a hipoxia tecidual, resultante do não desenvolvimento normal da vascularização retiniana, necessária para o metabolismo da retina, conduz à produção de factores angiogénicos com consequente processo de neovascularização. Esta poderá evoluir no sentido de proliferação fibrovascular anómala levando a tracção dos tecidos, descolamento da retina ou cegueira.
Vários factores de risco de RP têm sido apontados: baixo peso de nascimento, baixa idade gestacional, terapia com oxigénio (factor relevante, embora não constitua um pré-requisito), carência de vitamina E, exposição a produtos decorrentes de infecção/inflamação (como por ex., selectina-E, interleucinas), hiperglicémia, transfusões de sangue, RCIU, hipercápnia, anemia, hemorragia intraventricular, etc.. Dá-se grande importância ao papel dos factores de crescimento na génese da RP, designadamente IGF-1 e VEGF (vascular endothelial growth factor). O défice de síntese de IGF-1 no pré-termo seria responsável pela paragem do crescimento vascular retiniano.
A hipoxémia retiniana consecutiva à hipoperfusão origina síntese predominante de VEGF que se acumula no vítreo, sem efeito sobre o crescimento vascular. Uma vez retomada a secreção de IGF-I, associada à quantidade acumulada de VEGF, verifica-se intensa proliferação vascular retiniana. Quanto mais baixo o peso de nascimento e menor a idade gestacional, maior a probabilidade de, no recém-nascido pré-termo, se desenvolver retinopatia, em concomitância com elevadas probabilidades de complicações cardiovasculares e respiratórias.
Em suma, a RP parece, pois, ter uma relação directa com o grau de imaturidade vascular retiniana, acção do oxigénio sobre os vasos imaturos e factores que intervêm na oxigenação tecidual. Por outro lado, existem factores considerados protectores relativamente à mesma RP; entre eles contam-se a administração de esteróides no período pré-natal, e de surfactante ao recém-nascido (RN).
Classificação
O sistema classificativo de gravidade actualmente utilizado foi estabelecido pelo chamado CRYO-ROP Group, criado nos Estados Unidos da América do Norte. Como meio de estudo foi estabelecida a classificação, ainda em vigor. Muito do que se sabe hoje sobre a evolução da retinopatia e sua terapêutica deve-se aos estudos que este grupo tem efectuado, embora não seja de menosprezar o contributo doutros autores noutros países.
A classificação baseia-se fundamentalmente na extensão e localização da doença, assim como na sua gravidade. Classicamente são considerados os parâmetros que se seguem (Figura 3):
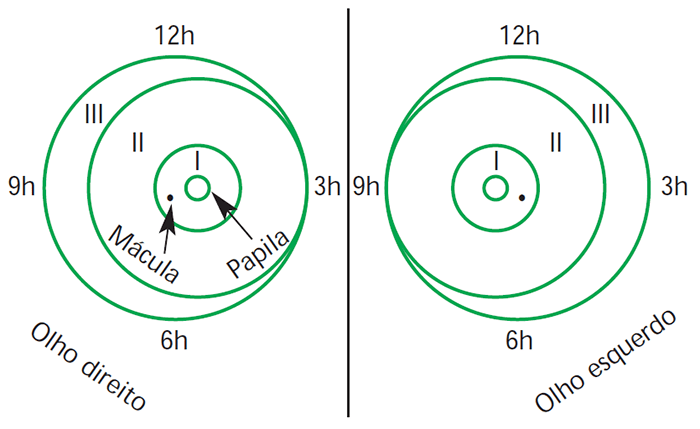
FIGURA 3. Camadas da retina: representação esquemática
Zona – Determina até que ponto progrediu o desenvolvimento da vascularização e onde residem as anomalias. Centrada no nervo óptico (papila ou disco óptico), a zona I compreende uma área circunferencial de raio duas vezes superior à distância papila-mácula lútea. É a porção mais posterior e a mais importante em termos de qualidade visual. A zona II é a área circunferencial distal à zona I, limitada anteriormente pela ora serrata nasal. A zona III corresponde ao crescente temporal remanescente.
Estádio – Indica a gravidade do processo, definindo-se pelo aspecto das alterações encontradas:
- A linha de demarcação separa a retina avascular da vascularizada;
- A linha de demarcação adquire volume, formando uma prega;
- Estendendo-se a partir da prega, observa-se tecido proliferativo fibrovascular extrarretiniano que se estende para o vítreo;
- Descolamento subtotal da retina; resulta da contracção do tecido proliferativo, que separa a retina da coroideia subjacente;
- Descolamento total da retina (corresponde à antiga designação fibroplasia retrolenticular).
Extensão – Designa a área circunferencial dentro da qual se observam alterações, expressas em sectores circulares de 30º (ou em “horas”); tem interesse apenas em estádios mais avançados: por ex., se a RP se estende entre 12 horas e 3 horas, terá extensão de 90º.
Doença plus (+) – Designa a existência de sinais de incompetência vascular. Traduz-se por dilatação e tortuosidade vascular progressivas que podem também atingir outras estruturas, como a íris. A presença de doença plus, habitualmente verificada em olhos que se encontram em estádio 3, conjugada como a sua extensão, pode sugerir a evolução iminente para um estádio de doença grave e irreversível, se não tratada.
A designação de doença rush denomina situações extremas em que há agravamento muito rápido, notório de dia para dia.
Nota: Cada caso é classificado em função do estádio mais avançado, da zona mais posterior e da presença de doença plus. Em regra, nos casos de RP ligeira até estádio 2, sem doença plus (+), verifica-se resolução completa.
Retinopatia limiar – Esta designação refere-se a estádio de evolução de retinopatia em que o risco de descolamento é ~ 50%; corresponde a uma retinopatia em estádio 3 atingindo a zona I ou II, numa extensão de 5 horas contínuas ou 8 horas descontínuas.
Retinopatia pré-limiar – Esta designação traduz um padrão requerendo acompanhamento mais rigoroso face ao risco de evolução para retinopatia limiar
Diagnóstico
O diagnóstico de RP baseia-se nos achados obtidos por oftalmoscopia binocular indirecta, da competência do oftalmologista.
Rastreio oftalmológico
Havendo antecedentes de prematuridade e factores de risco, torna-se obrigatório proceder ao rastreio da retinopatia aguda; tal rastreio deve começar ainda quando a criança está hospitalizada em unidades de cuidados intensivos neonatais.
De acordo com as normas de 2013 da Academia Americana de Pediatria e da Associação Americana de Oftalmologia Pediátrica e Estrabismo, são considerados lactentes em risco, por conseguinte com indicação para rastreio, todos aqueles com antecedentes de peso de nascimento (PN) inferior a 1500 g e de idade gestacional (IG) igual ou inferior a 32 semanas (ou com PN entre 1500 e 2000 gramas ou IG superior a 32 semanas havendo concomitantemente factores de risco, – designadamente instabilidade clínica e necessidade de suporte ventilatório).
O primeiro exame deve ser efectuado em função da IG ao nascer e da idade cronológica em semanas. (Quadro 3)
QUADRO 3 – Primeiro exame oftalmológico
| Idade Gestacional ao Nascer (semanas) | Idade Pós-natal (semanas) |
| 22 | 9 |
| 23 | 8 |
| 24 | 7 |
| 25 | 6 |
| 26 | 5 |
| 27-32 | 4 |
A partir do primeiro exame, o oftalmologista adapta o seguimento às particularidades de cada caso, designadamente as relacionadas com a gravidade.
No âmbito do rastreio da RP chama-se a atenção para uma aparelhagem moderna (câmara de fibras ópticas designada por RetCam, adaptada para fundoscopia e que permite visualizar imagens digitalizadas do fundo do olho com assistência por computador); é aplicável à telemedicina e pode ser utilizada por neonatologista treinado com o apoio de oftalmologia. Tal como para o método convencional, há necessidades de dilatação pupilar > 8 mm, o que se consegue com tropicamida a 0,5% (30-45 minutos antes da observação).
Estudos recentes em 2018, incidindo sobre os níveis do péptido natriurético urinário (NTproBNP) no RN em risco de retinopatia da prematuridade, concluíram que apenas nos casos com idade gestacional inferior a 30 semanas tal marcador poderá contribuir para identificar situação de risco de tal patologia.
A este propósito, especula-se que acima das 30 semanas, a patogénese da retinopatia poderá incluir certos mecanismos que poderão divergir dos verificados em idades gestacionais mais baixas.
Em termos de custo-benefício há que referir o custo de 100.000 dólares USA, confrontado com o custo dos cuidados assistenciais relacionados com a cegueira por RP: 1-5 milhões de dólares USA.
Após alta hospitalar devem ser feitos exames periódicos, regra geral entre os 6 e os 12 meses de idade pós-natal, entre os 2 e 2 anos e meio, entre os 3 e meio e 4 anos; e, depois, bianualmente.
Prevenção e tratamento
Na fase actual dos conhecimentos não existe qualquer fármaco para prevenir ou tratar a RP. Salienta-se, no entanto, como medida prioritária a prevenção do parto pré-termo, a necessidade de vigilância rigorosa da oxigenoterapia administrada ao RN pré-termo e da monitorização rigorosa da pressão arterial de O2 e de CO2 evitando a ultrapassagem de níveis críticos.
O objectivo do tratamento, da competência do oftalmologista, é deter a evolução para alterações estruturais graves (descolamento da retina), tentando evitar a perda visual, tendo em conta os efeitos iatrogénicos da própria terapêutica. Está indicado quando a retinopatia atinge o estádio limiar, ou pré-limiar com risco de descolamento de retina.
Através da ablação da retina isquémica, periférica ao tecido proliferativo, pretende-se diminuir a produção local de factores angiogénicos e deter o processo. A ablação é feita pela crioterapia (estudo CRYO-ROP) ou pelo método laser (árgon verde ou díodo vermelho). Pelas suas características menos agressivas, o método de fotocoagulação com laser de díodo é o método de eleição.
Na presença de descolamento de retina (estádios 4 ou 5) a ablação retiniana pode ser coadjuvada por vitrectomia e indentação escleral. No entanto, nesta fase o prognóstico é mau.
Nalguns centros tem sido utilizado um agente anti-angiogénico (à base de anticorpos inibidores de VEGF) intravítreo. Em 2011, de acordo com o estudo BEAT-ROP, foi demonstrada eficácia superior de bevacizumab/Avastin®. Admite-se que no futuro tenha papel importante o tratamento com células estaminais e com IGF-1.
Prognóstico
O período de tempo que medeia entre as 32 e as 42 semanas de gestação é crucial no que respeita à evolução da retinopatia. Pode ocorrer a sua regressão ou, pelo contrário, a evolução para formas graves. Na maioria dos casos ela regride, observando-se que o seu início ocorre por volta das 37 semanas de idade gestacional (entre as 34 e 46 semanas) e dura em média 15 semanas. A regressão com resolução completa é a regra nos casos de RP ligeira – estádio 1 ou 2, como foi referido antes.
No outro extremo de gravidade encontra-se a RP que atinge a zona I, situação associada a 90% de risco de progressão para descolamento da retina, se não tratada. Mesmo quando tratados, 50% de tais casos evoluem desfavoravelmente; por esta razão, mais recentemente tende a intervir-se mais precocemente nas crianças com RP na zona I ou na zona II.
Após a regressão espontânea ou induzida pelo tratamento, poderão manter-se alterações retinianas residuais de gravidade variável: em geral são tanto mais frequentes e mais graves quanto mais evoluído o estádio atingido.
Como consequência destas sequelas, são de destacar: o astigmatismo por distorção retiniana, a maculopatia cicatricial e o descolamento retiniano tardio do jovem ou adulto.
Os defeitos refractivos são mais frequentes nas crianças com antecedentes de prematuridade extrema (PN<1000 gramas). Mesmo não se tendo verificado evolução para RP grave, é mais frequente a ocorrência de miopia, astigmatismo e anisometropia (diferença significativa da capacidade refractiva entre os dois olhos). Este último defeito refractivo pode ser uma causa de ambliopia.
As perturbações da motilidade ocular (estrabismo e nistagmo) poderão tornar-se manifestas posteriormente, relacionando-se, quer com sequelas oculares (retinopatia e defeitos refractivos), quer com lesões do sistema nervoso central associadas.
Devido a erros refractivos significativos ou a estrabismo, a criança com antecedentes de prematuridade tem maior probabilidade de vir a ser amblíope.
Retinoblastoma
Aspectos epidemiológicos e importância do problema
O retinoblastoma, desenvolvendo-se a partir de células retinianas nucleadas imaturas, é o tumor maligno ocular mais frequente na criança. A célula estaminal ou primordial do retinoblastoma parece ser neuronal.
A sua incidência mundial oscila, de acordo com diversas estatísticas, entre 1 para 14.000 a 1/34.000 recém-nascidos. Em 90% dos casos surge antes dos 3 anos, sendo 30% bilateral. Numa minoria (10%) há antecedentes familiares.
A sua importância deriva essencialmente do facto de ser letal quando não tratado; inversamente, quando diagnosticado e tratado de forma oportuna, a percentagem de cura aumenta significativamente.
Etiopatogénese
O retinoblastoma representa a expressão fenotípica da ausência de um gene supressor tumoral, designado por gene do retinoblastoma ou RB1, que se localiza no braço longo, banda 14, do cromossoma 13 (13q14). Trata-se do primeiro gene supressor tumoral humano que foi completamente caracterizado. A sua função é suprimir o crescimento celular. Duas cópias normais do gene estão presentes na maioria das células humanas, sendo a sua função limitar o crescimento da célula; de referir que apenas uma cópia normal basta para cumprir a sua função. Antes de se conhecer a existência deste gene, o retinoblastoma era classificado como esporádico ou hereditário. Clínica e histologicamente, ambas as formas são indistinguíveis. A variedade hereditária, associando-se a tumores múltiplos e a compromisso binocular, pode ocorrer sob as formas autossómica dominante (mais frequente), ou recessiva.
Como resultado do crescimento celular superando a capacidade de irrigação sanguínea, surge um processo de necrose e de calcificação. Enquanto as células tumorais com origem nas camadas mais internas da retina crescem em direcção ao cristalino invadindo outras zonas da retina, as que se originam nas camadas mais externas podem levar ao descolamento da mesma. O retinoblastoma, através da invasão do nervo óptico ou da coroideia, pode invadir a órbita, salientando-se que a disseminação à distância, por via linfática ou sanguínea, surge raramente.
No respeitante ao tipo histológico em causa, cabe salientar que o sarcoma osteogénico é o mais frequente. Outros tipos tumorais possíveis são: neuroblastoma, condrossarcoma, rabdomiossarcoma, etc.. Nos casos de retinoblastoma hereditário existe elevada probabilidade de aparecimento de pinealoblastoma, altamente invasivo e letal, habitualmente ocorrendo nos primeiros quatro anos de vida.
Manifestações clínicas e diagnóstico
As principais formas de apresentação do retinoblastoma são estrabismo (em geral o primeiro sinal) e diminuição da visão ou leucocória. A propósito de leucocória, cabe referir a ausência do reflexo vermelho da pupila da criança quando o foco luminoso forte atravessa a pupila. (Figura 4)
Mais raramente e/ou com a progressão do tumor, este pode manifestar-se por hifema espontâneo (presença de sangue entre a íris e a córnea), glaucoma secundário, anisocória (midríase do olho afectado), heterocromia iridiana (diferente coloração da íris), nistagmo ou inflamação crónica. Em fases muito avançadas de proliferação ultrapassando os limites do globo, poderá sugerir o diagnóstico de celulite orbitária.

FIGURA 4. Leucocória do globo ocular esquerdo
De acordo com a classificação internacional (2007) são considerados cinco grupos de A a E em função da extensão: A (≤ 3 mm) ou small; B (bigger); C (contained); D (diffuse); E (extensive).
O diagnóstico diferencial faz-se, fundamentalmente, com a catarata, uveíte ganulomatosa anterior, toxocarose, toxoplasmose, retinite vírica, displasia da retina, retinosquise juvenil ligada ao X, etc., sendo que a anamnese e o exame objectivo geral se tornam fundamentais para orientar a destrinça.
O exame do fundo do olho sob dilatação pupilar, realizado por oftalmologista é fundamental; o aspecto é variável consoante o tipo de crescimento tumoral e a extensão e número de focos tumorais. (Figura 5)
A presença de calcificações retinianas em crianças com menos de 2 anos de idade é considerada sinal patognomónico de retinoblastoma, o que não acontece após os 2 anos. (Figura 6)
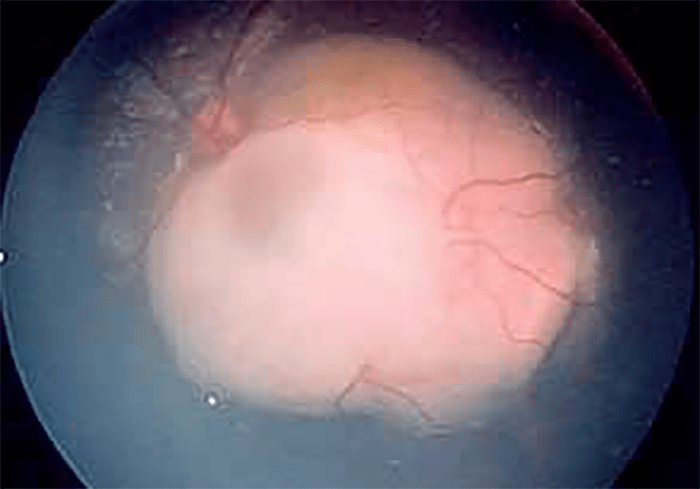
FIGURA 5. Retinoblastoma: aspecto da fundoscopia
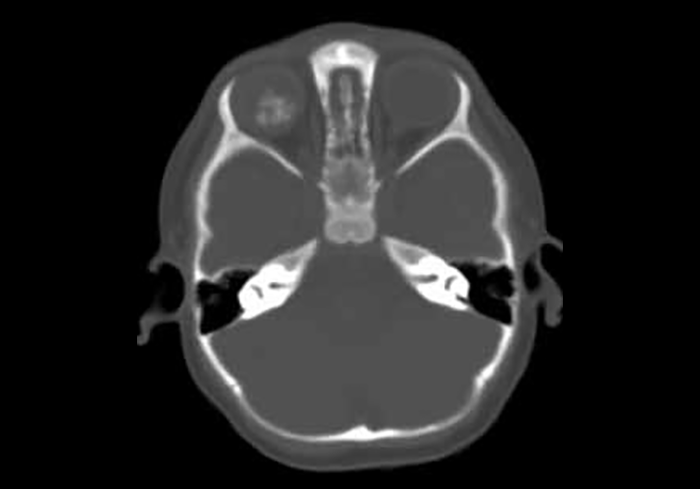
FIGURA 6. Retinoblastoma – imagem de calcificação (TAC)
Após fundoscopia, habitualmente repetida sob anestesia geral, o exame completa-se através da ecografia ocular realizada em ambos os olhos, e do estudo imagiológico (TAC, RM) para confirmação do diagnóstico e avaliação dos estádios evolutivos da doença.
Tratamento
O tratamento, da competência do oftalmologista, pode ser:
- conservador (nos casos de tumores de pequenas dimensões e intraoculares por ex. fotocoagulação, laser, termoterapia por laser díodo, quimiorredução, crioterapia, braquiterapia, radioterapia, etc.);
- radical (nos casos de grandes dimensões e associados à invasão do nervo óptico-enucleação). Salienta-se que a quimioterapia está indicada em associação às modalidades atrás referidas – a) e b).
Em centros especializados estão a ser usados novos tratamentos, alguns em fase experimental: vírus oncolíticos, genes “suicidas” tendo como vector adenovírus com gene de timidina de herpes simplex seguido de administração de ganciclovir, esterovírus angiostáticos, factor anti-VEGF, carboplatina de libertação lenta, etc..
Prognóstico
O factor de prognóstico mais importante é relacionável com o compromisso do nervo óptico; se forem detectadas células tumorais na margem do nervo óptico ou no espaço subaracnoideu, o prognóstico é mais reservado.
Nos casos de tumores unilaterais e intra-oculares, a taxa de cura é cerca de 90%.
O retinoblastoma bilateral associa-se a elevado risco de desenvolvimento doutras neoplasias primárias ao longo da vida, o que pode ainda ser potenciado pela radioterapia. O tempo médio de latência é cerca de 13 anos.
Nos casos de mutação genética germinal, o risco de recorrência é elevado.
Aconselhamento genético
O risco de retinoblastoma na descendência de um indivíduo com retinoblastoma só existe quando o doente tem uma mutação germinal. A avaliação do risco determina-se pela história familiar e pelo grau de compromisso tumoral, uni ou bilateral (ou multifocal). Os pais e irmãos de doentes afectados por retinoblastoma deverão também ser submetidos a exame fundoscópico. A penetrância do RB1 é cerca de 90%, o que corresponde a risco de passagem à descendência ~45%.
Agradecimento
As Figuras 4, 5 e 6 foram gentilmente cedidas pelos colegas Drs. Maria Araújo e Augusto Magalhães da Secção de Oftalmologia Pediátrica do Serviço de Oftalmologia do Hospital de São João, Porto, e a Figura 2 pelo Prof. João Goyri O´Neill da FCM/UNL e Serviço Universitário de Oftalmologia do Hospital Egas Moniz, Lisboa, a quem o editor e autora muito agradecem.
BIBLIOGRAFIA
Ahmadpour-K M, Jashni Motlagh A, Rasoulinejad SA, et al. Correlation between hyperglycemia and retinopathy of prematurity. Pediatr Int 2014; 56: 726-730
American Academy of Pediatrics, American Academy of Ophthalmology, American Association of Pediatric Ophthalmology and Strabismus. Screening examination of premature infants for retinopathy of prematurity. Pediatrics 2001; 108: 809-811
Aranda JV, Qu J, Valencia GB, Beharry KD. Pharmacologic interventions for the prevention and treatment of retinopathy of prematurity. Semin Perinatol 2019; 43: 360-366
Berrington JE, Clarke P, Embleton ND, et al. Retinopathy of prematurity screening at ≥30 weeks: urinary NTpro-BNP performance. Acta Paediatrica 2018; 107: 1722-1725
Brito C, Abrantes P. Oftalmologia. In Orientação Diagnóstica em Pediatria. Palminha J, Carrilho E M (eds). Lisboa: Lidel, 2003; 685-718
Cetinkaya M, Erener-Ercan T, Cansev M, et al. The utility of serial plasma sE-selectin measurements in the prediction of ROP in premature infants. Early Human Development 2014; 90: 517-521
Chiang MF, Keenan JD, Starren J, et al. Accuracy and reliability of remote retinopathy of prematurity diagnosis. Arch Ophthalmol 2006; 124: 322-327
Committee for the Classification of Retinopathy of Prematurity. Arch Ophthalmol 1984; 102: 1130-1134
Darlow BA, Ells AI, Gilbert CE, et al. Are we there yet? Bevacizumab therapy for retinopathy of prematurity. Arch Dis Child Fetal Neonatal Ed 2013; 98; F170-F74
Edwards EM, Horbar JD. Retinopathy of prematurity prevention, screening and treatment programmes. Semin Perinatol 2019; 43: 341-343
Gilbert C, Darlow BA. Retinopathy of prematurity. A world update. Semin Perinatol 2019; 43: 315-316
Good WV. Screening for retinopathy of prematurity: no ophthalmologist required? BJO 2000; 84: 127-128
Hartnett, ME. Pediatric Retina. Philadelphia: Lippincott Williams & Wilkins, 2005
Hong HK, Lee HJ, Ko JH, et al. Neonatal systemic inflammation in rats alters retinal vessels and stimulates pathologic features of ROP. J Neuroinflammation 2014; 11:87
Hutcheson KA. Retinopathy of prematurity. Curr Opin Ophthalmol 2003; 14: 286-290
Kanski JJ (ed). Oftalmologia Clínica. Barcelona: Elsevier, 2012
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Lee J, Dammann O. Perinatal infection, inflammation and ROP. Semin Fetal Neonatal Med 2012; 17: 26-29
Magalhães AA. Retina Pediátrica (Monografia). Porto: Sociedade Portuguesa de Oftalmologia, 2007
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Poll-Thé BT, Mailette L, Barth PG. The eye as window to inborn errors of metabolism. J Inherit Metab Dis 2003; 26: 229-244
Repka MX, Palmer EA, Tung B. Involution of retinopathy of prematurity. Cryotherapy for retinopathy of prematurity cooperative group. Arch Ophthalmol 2000; 118: 645-649
Reynolds JD, Dobson V, Quinn GE, Fielder AR, Palmer EA, Saunders RA, Hardy RJ, Phelps DL, Baker JD, Trese MT, Schaffer D, Tung B. Evidence-based screening criteria for retinopathy of prematurity: natural history data from the CRYO-ROP and LIGHT-ROP studies. Arch Ophthalmol 2002; 120: 1470-1476
Rodrigues P, Nepomuceno J, Brito C, Mesquita J. Perspectivas do estudo da diabetes ocular numa consulta de pediatria. Acta Pediatr Port 2003; 34: 13-15
VanderVeen DK, Cataltep SU. Anti-vascular endothelial growth factor intravitreal therapy for retinopathy of prematurity. Semin Perinatol 2019; 43: 375-380
Wu C, Petersen RA, VanderVeen DK. RetCam imaging for retinopathy of prematurity screening. JAAPOS 2006; 10: 107-111