Aspectos embriológicos e etiopatogénese
Pela 5ª semana da vida embrionária inicia-se uma fase de crescimento rápido do intestino médio (porção do tracto intestinal desde o duodeno até meio do cólon transverso). O intestino em crescimento dirige-se à cavidade celómica, e a respectiva porção distal liga-se ao canal onfalomesentérico.
Até à 10ª semana, continuando fora da cavidade abdominal do embrião, verifica-se aumento progressivo do comprimento do intestino, o qual é irrigado pela artéria mesentérica superior. A partir da 10ª semana o intestino reintroduz-se novamente na cavidade abdominal ao mesmo tempo que se verifica o processo de rotação que leva à sua fixação na parede abdominal posterior.
Não cabendo nos objectivos do livro uma descrição exaustiva do desenvolvimento embrionário do tubo digestivo, importa sintetizar que diversas perturbações verificadas neste processo podem ter várias consequências em termos de oclusão, ou de risco de oclusão, susceptível de se manifestar em diversos períodos da vida pós-natal. Eis alguns exemplos:
- Má rotação ou rotação incompleta;
- Atrésias intestinais explicáveis por diversos mecanismos, tais como: deformação de estruturas em desenvolvimento; acidentes vasculares intra-uterinos originando isquémia e necrose; volvo, encarceramento ou invaginação intestinais intra-uterinos;
- Ausência de células ganglionares nos plexos mioentéricos.
Manifestações clínicas e diagnóstico
Os principais dados da anamnese que poderão sugerir a existência de quadro de oclusão incluem: hidrâmnio, baixo peso de nascimento, vómitos biliosos, complicações pulmonares, presença ou ausência de mecónio e suas características, anomalias congénitas associadas, etc..
A realização de ecografia pré-natal permite o diagnóstico de oclusão intestinal em número significativo de casos.
O exame objectivo do recém-nascido realizado de modo sistemático permite igualmente a recolha de dados fundamentais salientando-se: pesquisa da permeabilidade esofágica e da permeabilidade anal utilizando procedimentos simples como a introdução de sondas; observação atenta do abdómen no sentido de detectar, quer aumento de volume ou distensão (indiciando, por exemplo, oclusão de grau variável do tracto digestivo inferior), quer depressão (sugestiva, por exemplo, hérnia diafragmática de Bochdaleck por ocupação torácica de vísceras abdominais ou atrésia do esófago sem fístula tráqueo-esofágica). Outros dados a pesquisar são: edema da parede, sinais de onfalite e a existência de circulação colateral.
Em suma, vómitos, distensão abdominal e ausência de dejecções/parésia intestinal, em graus variáveis, são sinais comuns nas diversas formas de oclusão. Os vómitos serão biliosos se a obstrução se localizar abaixo da ampola de Vater, e não biliosos se acima desta; a distensão abdominal é tanto mais acentuada quanto mais baixo o nível de obstrução; quando a distensão é muito acentuada, a elevação do diafragma pode originar dificuldade respiratória; por sua vez, a perda de secreções gástrica, biliar, pancreática e intestinal pode originar quadros de desidratação, choque hipovolémico, desequilíbrio hidroelectrolítico e ácido-base.
No que respeita a exames complementares indispensáveis, realça-se a radiografia abdominal simples (se possível em posição vertical) e a ecografia, os quais, na grande maioria das vezes permitem o diagnóstico.
Sistematização
Considerando os diversos quadros clínicos de oclusão do tracto digestivo numa perspectiva prática, neste capítulo procede-se a uma sistematização anatomofuncional das mesmas, com objectivo didáctico, no sentido craniocaudal (estômago, duodeno, jejuno-íleo e cólon-recto). Diversas entidades que se podem considerar abrangidas no conceito global de oclusão do tubo digestivo, como estenose hipertrófica do piloro, enterocolite necrosante e anomalias ano-rectais, integram capítulos específicos nesta Parte XXX do livro; o RGE e a doença de Hirschprung foram abordados em capítulos próprios.
ESTÔMAGO
Atrésia do piloro e outros defeitos do antro
Uma referência muito breve a uma situação rara – a atrésia do piloro – correspondendo (juntamente com outras anomalias obstrutivas do antro como “diafragmas” ou membranas) a 0,5% a 1% de todas as anomalias do tracto gastrintestinal. De acordo com a literatura, foram descritos casos familiares, admitindo-se transmissão hereditária autossómica recessiva. Tem sido associada à epidermólise bolhosa. Na maioria dos casos há antecedentes de poli-hidrâmnio. A ecografia pré-natal evidencia sinais de distensão gástrica.
As manifestações clínicas da atrésia do piloro são dominadas por distensão gástrica e vómitos não biliosos desde o primeiro dia de vida. Pela distensão gástrica impõe-se, pois, a aplicação de sonda gástrica para aspiração, obtendo-se como regra, > 20 mL de aspirado. Estão descritos casos de ruptura do estômago nas 1as 24 horas de vida. A ecografia e a radiografia abdominal simples feitas ao RN revelam sinais de distensão gástrica.
Nos casos de obstrução parcial (de grau variável) por membranas, o quadro manifesta-se mais tardiamente por vómitos, não progressão ponderal e dores abdominais. A endoscopia feita a crianças mais velhas permite evidenciar as pregas do antro.
O tratamento da síndroma de obstrução do antro gástrico inicia-se com a correcção do desequilíbrio hidroelectrolítico, desidratação e da alcalose hipoclorémica. Os vómitos persistentes obrigam a descompressão nasogástrica. Após estabilização do doente, procede-se à correcção (cirúrgica por laparotomia, ou por via endoscópica), em função do contexto clínico e idade.
Volvo gástrico
Este quadro verifica-se na sequência da torção do estômago sobre si mesmo superior a 180º; tal torção, que se pode concretizar segundo eixo longitudinal (volvo organoaxial) ou transversal (mesentérico-axial), resulta de ausência ou disfunção/hiperdistensão de determinados ligamentos de fixação gástrica (gastrofrénico, fazendo fixação segundo eixo transversal; e gastrosplénico, gastro-hepático e gastrocólico, segundo o eixo longitudinal).
Trata-se dum problema clínico raro, por vezes subdiagnosticado. Pode manifestar-se de forma aguda e crónica (esta última, mais frequente em crianças mais velhas); pode também estar associado a outros defeitos como má-rotação intestinal e asplenia.
As manifestações clínicas são inespecíficas, traduzindo-se por vómitos incoercíveis não biliosos e dor abdominal entre as refeições. O diagnóstico, uma vez suspeitado, obrigará a exames imagiológicos com contraste; verificam-se sinais de dilatação gástrica. Conforme o tipo de volvo, poderá observar-se sinal de nível líquido duplo com imagem “em bico” perto da junção gastresofágica no volvo mesentérico-axial, e de nível líquido simples sem o característico “bico” no volvo organoaxial.
O tratamento do volvo agudo constitui uma emergência cirúrgica, uma vez estabilizado o doente (gastropexia, precedida eventualmente por gastrostomia paliativa). Em casos seleccionados de volvo crónico, em doentes mais velhos (não lactentes), poderá estar indicado tratamento cirúrgico por via endoscópica. No pós-operatório está indicado tratamento médico anti-RGE.
Duplicação gástrica
Este defeito raro, explicável por falência da recanalização do intestino primitivo aquando do seu estádio “sólido, maciço ou acanalicular”, traduz-se pela existência de estruturas quísticas ou tubulares aderentes à parede interna do estômago (em geral de dimensões < 12 cm), em geral não comunicando com a cavidade gástrica. Em cerca de 35% dos casos há outros defeitos congénitos associados.
As manifestações clínicas são as de obstrução parcial ou completa da junção gastroduodenal (distensão gástrica e vómitos); nos casos de comunicação com a cavidade gástrica, poderão surgir ulceração, hematemeses e melenas. Por vezes, a estrutura anómala quística é palpável (~1/3 dos casos). Os exames de imagem (ecografia ou TAC) permitem esclarecer a situação clínica.
O tratamento consiste na excisão cirúrgica.
DUODENO
As oclusões localizadas no duodeno podem ser originadas por atrésia, estenose ou compressão extrínseca. Ao contrário das manifestações surgindo no contexto de oclusão jejunal ou ileal, nas oclusões duodenais não se verifica distensão abdominal e os vómitos não são biliosos (excepto quando o obstáculo é a jusante da ampola de Vater).
Atrésia e oclusão intrínseca e extrínseca do duodeno
Classificação e etiopatogénese
A oclusão do lume do duodeno pode ser completa ou incompleta, e de causa intrínseca ou extrínseca; de referir que poderão surgir diversos tipos de combinações das referidas modalidades de oclusão.
A oclusão incompleta ou parcial, de grau variável, surge como consequência de estreitamento ou estenose do lume duodenal e está, em geral, associada a compressão extrínseca do duodeno; pode ter várias causas:
- Bridas mesentéricas ou aderências peritoneais anómalas (bandas de Ladd) que acompanham situações de má rotação do cólon (oclusão extrínseca);
- Tecido pancreático aberrante, pâncreas anular, veia cava de localização pré-duodenal (oclusão extrínseca);
- Membrana ou diafragma parcialmente formados, ou fenestrados (oclusão intrínseca).
A causa mais frequente de compressão extrínseca é o pâncreas anular.
Na atrésia verifica-se oclusão total do lume duodenal, como resultado de anomalia do desenvolvimento embrionário (vacuolização incompleta do duodeno primitivo). Tal anomalia compreende três tipos:
- Diafragma ou membrana, completa e intacta (estrutura incluindo mucosa e submucosa);
- Cordão fibroso unindo dois “fundos de saco” os quais correspondem, respectivamente, aos segmentos proximal e distal do duodeno, sendo que o mesentério está intacto;
- Situação semelhante à anterior, mas sem cordão fibroso a unir os dois fundos de saco; neste tipo o mesentério está ausente.
Manifestações clínicas e diagnóstico
Nos exames imagiológicos pré-natais, em qualquer das situações atrás descritas, é possível detectar em cerca de um terço dos casos, presença de hidrâmnio associado a dilatação bolhosa gastroduodenal. A atrésia do duodeno está por vezes associada a outras anomalias do tubo digestivo, salientando-se a associação muito frequente a síndroma de Down (em cerca de 30% dos casos).
O quadro clínico pós-natal manifesta-se essencialmente por sinais de obstrução intestinal alta, isto é, com resíduo biliar gástrico volumoso e/ou esvaziamento gástrico demorado e incompleto; reitera-se, mais uma vez, a ausência de distensão abdominal.
No recém-nascido a presença de resíduo gástrico bilioso é sempre suspeita de oclusão duodenal. A eliminação de mecónio dependerá da verificação de oclusão completa ou incompleta e de lesões obstrutivas baixas associadas.
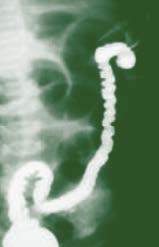
O estudo imagiológico a realizar com prioridade é o radiograma simples do abdómen em posição ortostática permitindo identificar o sinal característico e patognomónico da “dupla bolha” relacionável com oclusão completa/atrésia duodenal: a primeira “bolha” corresponde à distensão gástrica, e a segunda, à dilatação da primeira porção do duodeno. Por outro lado, não são observados sinais de “ar” nas ansas intestinais a jusante. (Figura 1)
Seguidamente, poderá proceder-se a estudo gastroduodenal, com contraste hidrossolúvel. Este estudo pode fornecer informações mais pormenorizadas sobre a arquitectura duodenal, o local da interrupção luminal ou a eventual presença de modelagem duodenal por compressão extrínseca. A Figura 2 mostra a imagem de distensão gástrica no contexto de atrésia da junção duodenojejunal. A ecografia abdominal poderá fornecer dados sobre a emergência dos vasos mesentéricos e a sua orientação no caso de má rotação intestinal; o estudo ecográfico da área pancreática pode fornecer dados sugestivos de pâncreas anelar.
Tratamento
O diagnóstico de oclusão duodenal implica sempre, qualquer que seja a anomalia em causa, uma abordagem cirúrgica correctiva.
A intervenção destina-se a tornar permeável o lume duodenal. Uma vez que cerca de oitenta e cinco por cento das oclusões duodenais têm como origem a região periampola, a correcção cirúrgica é realizada por meio de uma derivação a esse ponto por duodenoduodenostomia laterolateral.
No caso de oclusão intrínseca incompleta pode ser realizada uma duodenotomia seguida de exploração endoluminal e excisão do obstáculo mucoso, quer seja um diafragma fenestrado, quer seja uma manga (wind-sock). Nos casos de compressão extrínseca, deverão ser libertadas todas as aderências peritoneais anómalas presentes. Na impossibilidade de retirar o obstáculo extrínseco, a derivação duodenal deverá ser construída mais proximalmente, com uma verdadeira derivação “by-pass” ao arco duodenal, por meio de uma gastroenterostomia laterolateral. Como principais complicações da derivação duodenal citam-se a deiscência da anastomose duodenal e a estenose cicatricial.
Seguimento
Pelas razões apresentadas anteriormente, a derivação duodenal implica a instituição de pausa alimentar, aspiração gástrica activa e nutrição parentérica total, durante um período ~ 10-14 dias. Após este período é introduzida a nutrição entérica, cuja progressão em concentração e quantidade, é feita de acordo com a tolerância demonstrada pelo doente.
Prognóstico
O prognóstico das situações de oclusão duodenal é na generalidade excelente na ausência de complicações cirúrgicas.
O prognóstico definitivo depende da eventual associação doutras anomalias, nomeadamente cardíacas.
JEJUNO E ÍLEO
Atrésia e estenose do jejuno e íleo
Classificação e etiopatogénese
Atrésia e estenose jejunoileal são defeitos congénitos em que se verifica, respectivamente, a obliteração completa ou parcial do lume intestinal no segmento respectivo.
A atrésia é responsável por cerca de um terço dos casos de oclusão intestinal no recém-nascido. A distribuição por sexos é similar, oscilando a frequência entre 1/1.300 a 1/5.000 nados-vivos.
A etiopatogénese de tais anomalias relaciona-se provavelmente com perturbações de vascularização e fenómenos isquémicos mesentérico-intestinais dando origem a défice da permeabilidade do intestino primitivo; tais alterações parecem explicar igualmente defeitos mesentéricos associados.
As atrésias jejunoileais são classificadas em quatro tipos:
Tipo 1: obliteração luminal por membrana com continuidade da parede e mesentério normal (cerca de 30%);
Tipo 2: cordão fibroso unindo os topos proximal e distal do intestino, em fundos de saco, sendo que o mesentério é normal (cerca de 25%);
Tipo 3a: semelhante ao tipo 2, sem cordão fibroso e fundos de saco separados; associado a defeito mesentérico em “V” (cerca de 15%);
Tipo 3b: obliteração luminal proximal e defeito mesentérico e vascular do território distal, sendo este vascularizado por um único vaso em circulação retrógada (apple-peel deformity ou atrésia em forma de árvore de Natal) (11%);
Tipo 4: múltiplas atrésias (cerca de 17%).
A atrésia jejunoileal pode estar associada a outras anomalias tais como síndroma de Down, defeitos cardíacos, a associação VACTERL, doença de Hirschsprung, gastrosquise e íleo meconial.
Manifestações clínicas e diagnóstico
A ecografia pré-natal pode evidenciar sinais de hidrâmnio e de distensão gástrica fetal. Os sinais clássicos no recém-nascido são: vómitos biliosos, ausência de mecónio e distensão abdominal, tanto mais acentuada quanto mais distal o segmento em que se verifica a oclusão.
A radiografia simples do abdómen (realizada idealmente em posição vertical) evidencia sinais de ansas intestinais dilatadas com ou sem níveis hidroaéreos. (Figuras 3 e 4) Quando estes sinais são muito exuberantes, no diagnóstico diferencial haverá que incluir a doença de Hirschprung (Figura 5) e o íleo meconial. Em função do contexto clínico, poderá estar indicado o clister opaco.
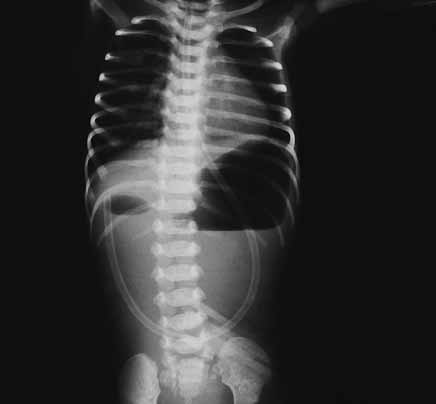
FIGURA 1. Dupla bolha: Sinal radiológico de oclusão duodenal (completa). (URN-HDE)
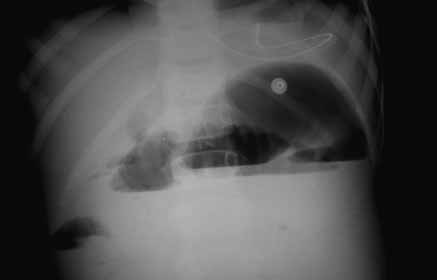
FIGURA 2. Distensão gástrica por atrésia da junção duodenojejunal. Ausência de ar a jusante da zona de atrésia (radiografia tóraco-abdominal). (NIHDE)
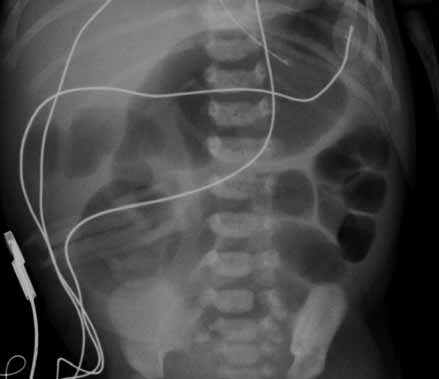
FIGURA 3. Imagem de radiografia simples abdominal evidenciando distensão acentuada de ansas do jejuno no contexto de atrésia ileal. (UCIN-HDE)
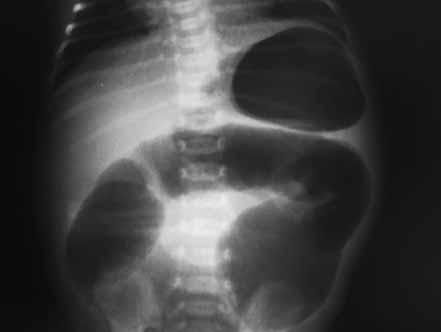
FIGURA 4. Atrésia jejunoileal; imagem de radiografia simples do abdómen evidenciando distensão “gigante” de ansas do jejuno parecendo distensão cólica. (UCIN-HDE)
FIGURA 5. Oclusão intestinal baixa, evidenciando níveis hidroaéreos.
Tratamento
Uma vez confirmado o diagnóstico, está indicada intervenção cirúrgica cujo objectivo é promover a continuidade do trânsito intestinal, procedendo a anastomose digestiva directa, após remodelar o segmento dilatado. Se houver sinais de necrose intestinal, procede-se a ressecção da ansa afectada. Pressupõe-se a realização dum conjunto de cuidados pré-operatórios que dizem respeito, essencialmente a aspiração nasogástrica, e manutenção do equilíbrio hemodinâmico, hidroelectrolítico e ácido base.
É habitual surgir no período pós-operatório disfunção anastomótica resultante dos diferentes calibres de ansa, do tipo de sutura, da forma da anastomose, e da alteração da motilidade intestinal associada ao segmento pré-atrésia. Por estas razões, os doentes com tal patologia permanecem durante um período variável de tempo dependentes exclusivamente da nutrição parentérica veiculada, de preferência, através de cateter central de longa duração de tipo Hickman-Broviac.
Os aspectos chave do período pós-operatório são a aspiração nasogástrica activa e a nutrição parentérica. As complicações são decorrentes do tipo de atrésia encontrada, da exequibilidade ou não de reconstituição do trânsito intestinal e da técnica cirúrgica em si. Neste tipo de anomalia surge invariavelmente período mais ou menos prolongado de pseudo-obstrução intestinal com resíduo gástrico abundante que pode ser resultante de dismotilidade intestinal, designadamente.
Se se verificar deiscência anastomótica, torna-se obrigatória a reintervenção cirúrgica imediata. A existência de uma obstrução mecânica evidente por estenose cicatricial ou angulação da anastomose obriga também a efectuar uma revisão cirúrgica da situação.
Prognóstico
O prognóstico global destes doentes (cuja sobrevida é > 95%) é ditado pela precocidade do diagnóstico, pela presença de anomalias congénitas associadas, da imaturidade, da eventual necessidade de ventilação assistida prolongada, das complicações associadas ao tipo de atrésia intestinal, e da técnica cirúrgica utilizada.
Todo este quadro é agravado pela presença de atrésias de tipo 3 ou 4, complicadas de encurtamento intestinal, podendo originar síndroma de intestino curto.
Íleo meconial
Definição e etiopatogénese
Define-se íleo (ou ileum) meconial como a situação clínica de oclusão ileal distal por mecónio anormal, espesso e viscoso, devida a mucoviscidose/fibrose quística; em cerca de 10% dos casos é a primeira manifestação desta doença. A oclusão (intraluminal), surgindo em cerca de 10% a 20% dos recém-nascidos com tal doença, verifica-se na região pré-valvular (válvula ileocecal) numa extensão de cerca de 15 a 20 cm; a montante desta zona verifica-se dilatação do íleo.
Com efeito, na fibrose quística todas as glândulas secretoras de muco são anormais, sendo de referir que, para a anormalidade do mecónio, contribuem a insuficiência de enzimas pancreáticas proteolíticas e a secreção de mucoproteínas anormais pelas células caliciformes do intestino delgado; de facto, o mecónio destes doentes contém menor concentração de hidratos de carbono e maior de proteínas; a proteína mais abundante é a albumina, com uma concentração 5 a 10 vezes superior ao normal, o que explica a sua extrema viscosidade.
Manifestações clínicas e diagnóstico
A apresentação clínica no período neonatal é caracterizada por distensão abdominal, resíduo gástrico, vómito de características biliosas e ausência de emissão de mecónio nas primeiras 48 horas de vida. A palpação abdominal permite delimitar, por vezes, as ansas distendidas, assim como massa depressível correspondendo ao mecónio espesso impactado. O ânus e recto têm calibre reduzido face à condição de microcólon de desuso.
O exame radiográfico abdominal simples permite demonstrar sinais de distensão intestinal do delgado, ausência de níveis hidro-aéreos e presença de imagens de “bolha de sabão” ou “vidro despolido” traduzindo a mistura gasosa e meconial no território ileal distal (quadrante inferior direito do abdómen).
O clister opaco demonstra a existência de microcólon de desuso (calibre muito estreito) devido à obstrução ileal distal, por vezes com presença de pequenas concreções meconiais mais espessas no cólon proximal e íleo distal.
As formas complicadas traduzem-se fundamentalmente por distensão abdominal progressiva, dificuldade respiratória, perfuração e peritonite no período pré-natal; igualmente poderão existir: sinais de compromisso de ansa intestinal como torção mesentérica e compromisso isquémico, volvo e/ou atrésia, efeito de massa sobre as ansas intestinais pela presença de um quisto meconial, e calcificações intra-abdominais secundárias a peritonite meconial pré-natal.
Diagnóstico diferencial
Com raras excepções, a situação compatível com íleo meconial, até prova em contrário, pode considerar-se um epifenómeno da fibrose quística. No entanto, haverá que atender às seguintes situações:
- A fibrose quística pode manifestar-se no recém-nascido por atraso de eliminação de mecónio ou por eliminação de rolhão meconial espesso com oclusão transitória do cólon distal;
- A chamada síndroma do rolhão meconial (situações de mecónio espesso de etiopatogénese diversa da associada à fibrose quística e mais frequente em recém-nascidos de baixo peso) poderá originar um quadro clínico semelhante ao íleo meconial propriamente dito (associado à fibrose quística).
Classificação e tratamento
O íleo meconial classifica-se em simples e complicado consoante a seu modo de apresentação e a sua resolução terapêutica (ver atrás).
- O íleo meconial simples é tratado de forma não cirúrgica, por meio de clister de substâncias que se destinam a dissolver o mecónio impactado, favorecendo a sua expulsão por via rectal (gastrografina e acetilcisteína).
A gastrografina é uma solução aquosa de diatrizoato de metilglucamina que, por mecanismo osmótico, promove a transferência de água no sentido células intestinais → lume intestinal, diminuindo a viscosidade do mecónio. A acetilcisteína é uma enzima proteolítica que promove a liquefação do mecónio, sendo em geral usada após o clister de gastrografina.
- Nos casos de íleo meconial complicado (integrando situações atrás descritas, em que não é possível resolução pelo método de tratamento conservador), existe sempre indicação cirúrgica.
– Nas formas sem compromisso de ansa intestinal, é realizada uma enterotomia para irrigação endoluminal com o objectivo de dissolver localmente o mecónio impactado.
– Nas formas de apresentação com compromisso de ansa, isto é complicadas de torção de mesentério, volvo, perfuração in utero ou formação de peritonite meconial ou quisto meconial intra-abdominal, é necessário realizar uma ressecção segmentar do segmento afectado e, posteriormente, restabelecer a continuidade intestinal, ou derivar temporariamente o intestino, encerrando a enterostomia em segundo tempo cirúrgico.
A abordagem cirúrgica do íleo meconial obriga também à colocação de um cateter central de longa duração do tipo Hickman-Broviac para permitir a administração de nutrição parentérica.
Complicações pós-operatórias
As complicações pós-operatórias precoces mais frequentes resultam da enterotomia realizada para a irrigação endoluminal e da anatomose pós-ressecção segmentar de ansa que pode ser complicada por deiscência ou por obstrução mecânica.
As complicações tardias são devidas essencialmente a alterações da motilidade do segmento ileal distal obrigando, por vezes, à instituição de fármacos pró-cinéticos.
As complicações a longo prazo resultantes, sobretudo, da ressecção do segmento ileal distal, derivam da alteração do ciclo êntero-hepático e da necessidade de nutrição parentérica de longa duração: litíase biliar e doença hepática colestática.
Por fim, haverá que equacionar outras complicações inerentes à doença de base – a fibrose quística.
Seguimento e prognóstico
O seguimento destes doentes é de extrema importância e deverá ser efectuado em centros especializados dispondo de equipa multidisciplinar.
O prognóstico no primeiro ano de vida é decorrente da forma de apresentação da doença e do sucesso das opções terapêuticas tomadas. A sobrevivência no primeiro ano de vida nos casos não complicados é > 95% e, nos casos complicados, ~90%.
Má-rotação
Definição e etiopatogénese
A má rotação intestinal consiste num defeito de rotação e de fixação (não fixação), na cavidade peritoneal, da ansa primitiva em torno do eixo vascular que origina a artéria mesentérica superior. Esta anomalia integra, pois, também um componente vascular; tal explica a possibilidade de ocorrência concomitante de complicações graves resultantes de isquémia intestinal que podem surgir nos casos de má rotação complicada de volvo do intestino médio.
Trata-se dum problema clínico, com muitas variantes anatómicas, que pode ser assintomático; as formas sintomáticas, manifestando-se na sua maioria até ao 1 ano de idade (em especial no RN) surgem na proporção aproximada de 1/7.000 RN. Outros defeitos congénitos associados a má rotação incluem com maior frequência: atrésia duodenojejunal, onfalocele, gastrosquise e hérnia diafragmática.
Na má rotação completa (não rotação ou verdadeira má rotação) a totalidade do cólon e o íleo terminal localizam-se no lado esquerdo do abdómen, enquanto o duodeno e jejuno se situam no lado direito.
Existe um mesentério comum, não fixado à parede posterior abdominal, sendo que o cego se localiza nos quadrantes superiores ou em posição aproximada do centro do abdómen. Poderá verificar-se a existência de pregas ou fitas de peritoneu (as chamadas bandas ou bridas de Ladd) entre o cego e a parede póstero-lateral do abdómen, cruzando e comprimindo o duodeno, o que causa oclusão; a montante das bridas o duodeno está dilatado e, a jusante, atrófico. O íleo terminal está colado ao jejuno proximal por aderências ou bridas peritoneais anormais; esta anomalia de posição cria um pedículo intestinal estreito, o que predispõe a volvo intestinal (enrolamento ou torção sobre si mesmo ou em roda de ponto fixo – por não fixação do intestino –, com consequente oclusão e perturbação circulatória isquémica).
Outras variantes da chamada má rotação incluem as rotações incompletas e as fixações incompletas.
Manifestações clínicas e diagnóstico
As manifestações clínicas desta entidade podem ser muito variáveis.
A forma de apresentação mais frequente traduz-se por vómitos biliosos intermitentes no período neonatal, sugerindo obstrução duodenal. (Figura 6)*
A forma de apresentação mais grave é o volvo** do intestino médio, por vezes a primeira manifestação da anomalia: agravamento abrupto do estado geral com distensão abdominal, dores abdominais/cólicas no lactente, irritabilidade e, por vezes, eliminação de fezes com sangue, e sinais de choque hipovolémico; este quadro constitui uma emergência.
O exame físico poderá evidenciar ausência de distensão abdominal, ou distensão muito discreta nos casos de localização alta da obstrução.
A ocorrência de vómitos biliosos constitui, em geral, o evento que desencadeia a investigação etiológica. Perante a suspeita clínica de quadro oclusivo intestinal, a radiografia abdominal simples (realizada sempre como primeira prioridade para o diagnóstico) poderá revelar sinais de distensão acentuada de ansas (Figura 7) e, eventualmente, o sinal da “dupla bolha”, patognomónico da oclusão duodenal que, como foi referido, poderá ser um acompanhante da má rotação.
Se a radiografia simples do abdómen evidenciar sinais de duodeno dilatado e de presença de gás nos quadrantes inferiores do abdómen, está indicada a realização de trânsito gastroduodenal contrastado com bário, exame que permite demonstrar a posição do duodeno, a sua forma, e a localização do ângulo de Treitz. Nos casos de má-rotação, o duodeno tem uma forma espiralada, sem se verificar a sua curvatura harmoniosa para a esquerda, e o ângulo de Treitz não está definido no hipocôndrio esquerdo.
O clister opaco pode dar uma imagem indirecta de má-rotação pela posição anómala do cego, que geralmente se encontra em posição elevada nos quadrantes direitos do abdómen ou em posição central.
*Alta da maternidade às 48 horas de vida. Reinternamento aos 4 dias de vida por vómitos biliosos e intolerância alimentar progressiva. A laparotomia comprovou má-rotação de 270º, tendo sido realizada desrotação anti-horária, libertação do ângulo de Treitz, e bipartição do mesentério (operação de Ladd Gross). (caso clínico do Dr. Rui Alves) **Recorda-se a definição de volvo (ou vólvulo): enrolamento ou torção de um órgão oco sobre si mesmo ou em torno de um ponto fixo, tendo como consequência obstrução e perturbações isquémicas graves por compromisso circulatório local. |
A ecografia abdominal na sua variedade de doppler poderá evidenciar dados indirectos quanto à origem e direcção dos vasos mesentéricos, nomeadamente o sinal doppler em turbilhão (Whirlwind sign), típico da má-rotação intestinal.
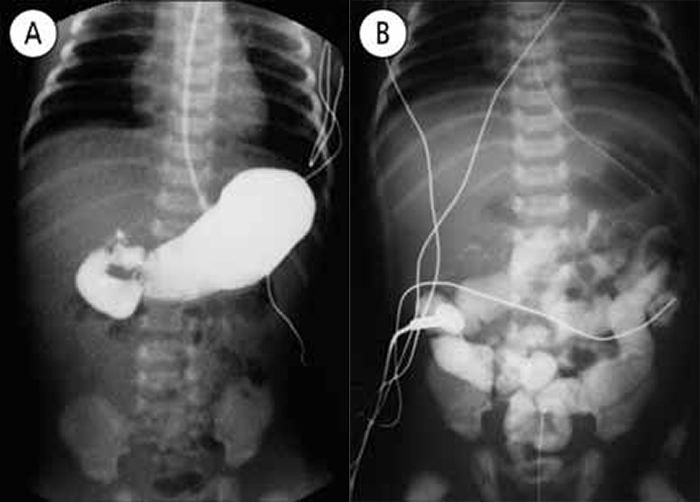
FIGURA 6. Caso de obstrução intestinal alta. Vómitos alimentares alternando com períodos de boa tolerância alimentar. Imagem radiográfica tóraco-abdominal com contraste introduzido no estômago. (UCIN-HDE)*
A – Aparente posição normal da 1ª e 2ª porção do duodeno com interrupção do contraste a jusante da 2ª porção, sugerindo possível obstrução ao nível do ângulo de Treitz; B – Verificação de passagem livre do contraste cerca de 1 hora após radiografia A.
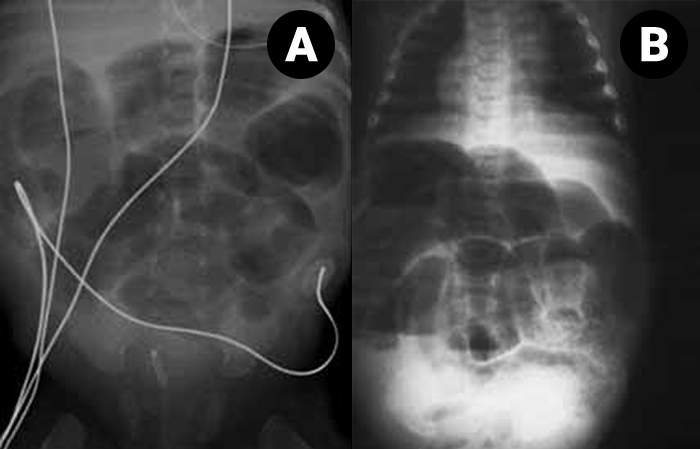
FIGURA 7. Volvo do intestino médio: sinais de distensão de ansas (radiografia abdominal simples. (UCIN-HDE)**
Tratamento
A chave do sucesso terapêutico é o elevado índice de suspeita diagnóstica para uma decisão rápida quanto à correcção cirúrgica, obrigatória. Com efeito, o atraso na obtenção do diagnóstico e na decisão terapêutica pode acarretar a perda extensa de segmentos intestinais por necrose isquémica; por outro lado, a correcção cirúrgica da situação contribui para a prevenção do volvo do intestino médio.
Como medidas gerais pré-operatórias são referidas a manutenção do equilíbrio hemodinâmico, hidro-electrolítico e ácido base, assim como a aplicação de sonda gástrica para descompressão do estômago e da primeira porção do duodeno dilatada.
A técnica cirúrgica utilizada (operação de Ladd-Gross cuja descrição ultrapassa o âmbito deste livro), essencialmente permite desfazer a rotação intestinal e libertar as “bandas de Ladd” e aderências peritoneais em geral.
Nos casos de volvo do intestino médio em que se pode verificar compromisso isquémico, muitas vezes irreversível, do território irrigado pela artéria mesentérica superior, o procedimento cirúrgico descrito, destina-se também a realizar a desrotação mesentérica e a permitir a perfusão terminal das ansas intestinais.
Prognóstico
O seguimento destes doentes, nos casos não complicados, não implica qualquer cuidado especial, quer sob o ponto de vista nutricional, quer sob o ponto de vista do desenvolvimento, uma vez que a cirurgia se pode considerar, em princípio, curativa. Contudo, em cerca de 10% dos casos, poderá verificar-se manutenção da sintomatologia obstrutiva no período pós-operatório imediato ou mais tardiamente; tal sintomatologia pode explicar-se por recorrência de torção parcial mesentérica, por bridas ou aderências, ou por dismotilidade intestinal.
Em cerca de 15% dos casos poderá surgir perda intestinal extensa por necrose isquémica secundária a volvo do intestino médio, conduzindo ao quadro de síndroma de intestino curto.
Nos casos de perda intestinal por isquémia (mais ou menos extensa), o prognóstico depende igualmente da qualidade funcional dos segmentos intestinais remanescentes e do respectivo capital de regeneração intestinal.
A mortalidade associada a esta anomalia varia entre 3% e 9% estando invariavelmente associada à ocorrência de volvo do intestino médio, à prematuridade e à extensão da necrose intestinal.
Invaginação intestinal
Definição
A invaginação intestinal é uma situação clínica resultante da penetração de um segmento proximal do intestino (intussusceptum) – como um telescópio ou à maneira de um dedo de luva do avesso – noutro segmento do intestino mais distal, que o recebe (intussuscepiens). (Figura 8)

FIGURA 8. Representação esquemática do mecanismo da invaginação intestinal. (consultar texto)
Aspectos epidemiológicos
Este problema clínico surge geralmente entre os 4 e os 10 meses, com um “pico” aos 7 meses, e limites entre os 3 meses e os 3 anos. O sexo masculino é cerca de 3 vezes mais afectado do que o feminino. O primum movens (ou “cabeça de invaginação”) desta mobilidade anómala do intestino poderá ser a hiperplasia linfóide (protusão para o lume do intestino das placas de Peyer, relacionada com infecção vírica), o que é demonstrado em cerca de 50% dos casos nalgumas séries.
Trata-se da causa mais frequente de obstrução intestinal no grupo etário atrás referido; a localização mais frequente é a íleo-ceco-cólica, o que é explicável pela maior riqueza de placas de Peyer nesta região do intestino.
No recém-nascido há que admitir possível duplicação intestinal como factor causal da invaginação (ver adiante). Na criança com mais de 3 anos é muito provável que haja certas lesões que sirvam de “cabeça“ da invaginação, tais como: divertículo de Meckel, apêndice ileocecal, pólipos, tumores carcinóides, lesões hemorrágicas da púrpura de Henoch-Schonlein, linfoma não Hodgkin, corpos estranhos, pâncreas ectópico ou mucosa gástrica ectópica. A incidência de lesões anatómicas que funcionam como “cabeça de invaginação” aumenta com a idade.
Manifestações clínicas
A anamnese, em geral, só por si, permite o diagnóstico. Na sua forma típica, no lactente, em plena saúde verifica-se início de um episódio de cólicas abdominais intensas e mal-estar, (traduzido por episódios de “dobrar” os membros inferiores sobre o abdómen de forma aflitiva), por vezes associado a vómitos, palidez e sudação intensa. O episódio, com a duração de alguns minutos, é intercalado por pausas de acalmia em que o bébé fica apático ou letárgico. Ao cabo de alguns minutos da referida acalmia, a aparência de dor e os restantes sinais voltam de novo e de modo súbito. Por vezes há emissão de fezes normais a que se segue, numa fase mais avançada, a emissão de fezes tingidas de sangue e, mais tarde, já só coágulos mucóides de cor vermelha escura exibindo o típico aspecto de “geleia de framboesa“.
O passo mais importante do exame objectivo é a palpação abdominal durante a qual é possível, na forma mais habitual – a invaginação íleo-cecocólica – encontrar a fossa ilíaca direita “vazia” (pois o cego subiu) e palpar massa em “chouriço” no hipocôndrio direito (correspondente à zona invaginada). Nas formas mais avançadas é possível que o intestino invaginado surja exteriorizado pelo ânus. Procedendo-se ao toque rectal torna-se possível palpar a cabeça da invaginação com o dedo explorador, o qual sairá “sujo” de sangue.
Existe uma forma especial de invaginação (invaginação intestinal pós-operatória, na sua grande maioria íleo-ileal) que pode surgir na sequência de intervenções cirúrgicas abdominais muito invasivas: manifesta-se cerca de 2 semanas depois da intervenção cirúrgica, essencialmente por distensão abdominal, vómitos biliosos e sinais de estase gástrica crescente.
Exames complementares
O exame de eleição para o diagnóstico é a ecografia; em caso de invaginação intestinal, a mesma revela sinais de duplo contorno do intestino invaginado que se traduz na clássica “imagem em alvo” (Figura 2 do Capítulo sobre Imagiologia na Parte II). Nalguns centros é utilizada a ecografia de modo contínuo durante um período de 24 horas.
A radiografia simples do abdómen mostra sinais de oclusão intestinal e o clister opaco permite a localização. Este último (sempre indicado excepto nos casos em que se verifiquem sinais de irritação peritoneal) poderá igualmente ter efeito terapêutico. De facto, se se elevar o frasco de contraste baritado com que se realiza o clister até um máximo de 70 cm acima do plano do doente em decúbito, poderá assistir-se à resolução do problema: desinvaginação causada pela pressão hidrostática da coluna de bário.
Tratamento
Perante uma suspeita de invaginação intestinal, a primeira atitude deve ser a introdução de tubo nasogástrico para aspiração e o estabelecimento de linha endovenosa de fluidoterapia para correcção da eventual desidratação relacionada com perdas por vómitos e para o terceiro espaço.
A ecografia poderá ser realizada antes de corrigido o desequilíbrio hidroelectrolítico.
Como foi antes referido, o clister opaco é, muitas vezes, terapêutico. Este procedimento deverá ser realizado com a presença do cirurgião; a eficácia do mesmo (desinvaginação) pode ser comprovada pela verificação do refluxo do contraste do cego para o íleo terminal, através da válvula ileocecal. Refira-se, no entanto, que este critério não é obrigatório, pois em cerca de 1/3 dos indivíduos a válvula ileocecal é continente.
Uma boa alternativa ao clister opaco convencional será a desinvaginação pneumática (introdução de ar sob pressão controlada com um esfigmomanómetro, em alternativa ao contraste baritado.
A intervenção cirúrgica está indicada quando se verificar:
- Sintomatologia sugestiva de irritação peritoneal;
- Obstrução intestinal;
- Falência do clister opaco ou pneumático;
- Recorrência de invaginação (a partir da terceira crise após 2 desinvaginações eficazes).
Durante a intervenção cirúrgica procede-se à desinvaginação manual por expressão cautelosa da ansa invaginada (e não por tracção que pode levar à ruptura), à ileocecopexia quando indicada, e à ressecção de segmento intestinal em caso de perfuração.
Quistos enterogénicos (Duplicação intestinal)
Definição e importância do problema
A chamada duplicação intestinal é uma anomalia tumoral quística ou tubular que faz parte, sob o ponto de vista da etiopatogénese, dum defeito mais vasto, com localização variável, desde a boca ao ânus (duplicação do tracto gastrintestinal); o local mais frequente de aparecimento da duplicação é o intestino delgado, principalmente o íleo.
Em exames necrópsicos a frequência apurada é cerca de 1/5.000.
Etiopatogénese
Segundo a teoria mais consensual sobre a etiopatogénese da duplicação intestinal, este defeito forma-se do seguinte modo: até cerca da 7ª semana de gestação o intestino tem forma cilíndrica maciça o que se deve à proliferação epitelial; a partir desta fase, ocorre um processo de vacuolização central (vacúolos interligando-se e comunicando) que leva a que o referido “cilindro maciço” se transforme em “tubo”; quando alguns vacúolos não se fundem, formam-se estruturas quísticas adjacentes ou duplicação “do tubo”, ocorrendo, por vezes, em mais de um segmento.
Reportando-nos à localização intestinal, o referido quisto localiza-se no respectivo bordo paramesentérico, compartilhando a irrigação sanguínea e evidenciando o mesmo epitélio do intestino adjacente. Em cerca de 30% dos casos o epitélio é de tipo gástrico, do que resulta a possibilidade de acumulação de secreção gástrica intraquística por deficiente drenagem, com inflamação, hemorragia e/ou perfuração consequentes.
Manifestações clínicas e diagnóstico
Na maioria dos casos, as manifestações surgem nos primeiros dois anos de vida, dependendo os sinais e sintomas da localização e das dimensões do defeito estrutural; de salientar que as duplicações de pequenas dimensões poderão ser assintomáticas.
As anomalias mais frequentemente associadas são: vertebrais, má rotação intestinal e nefrourológicas.
A tríade clássica (melena, hemorragia e massa abdominal móvel) surge nalgumas séries com uma frequência ~50%. Nos casos de duplicações jejuno-ileais os quadros inaugurais (de oclusão) poderão ser invaginação intestinal ou volvo.
No âmbito da vigilância pré-natal a ecografia pode identificar a anomalia.
Sempre que se suspeita de duplicação intestinal estão indicados exames imagiológicos. A ecografia constitui o exame de primeira linha; sempre que esta não é esclarecedora, deve proceder-se a tomografia axial computadorizada.
Nos casos de hemorragia digestiva, a cintilografia poderá ter utilidade para pesquisa de mucosa gástrica ectópica.
Cabe referir, a propósito, que na investigação de duplicações com outra localização estão indicados: estudo do trânsito gastrintestinal com contraste; endoscopia digestiva alta (estômago e duodeno); e clister opaco (cólon e recto).
Tratamento
O tratamento das duplicações do tracto gatrintestinal é cirúrgico, procedendo-se a ressecção completa pelo risco de desenvolvimento ulterior de neoplasia.
CÓLON E RECTO
A atrésia do cólon, mais frequente no cólon transverso, é muito rara, correspondendo a cerca de 6% das atrésias intestinais em geral. As atrésias múltiplas no cólon são também extremamente raras.
Doença de Hirschprung (megacólon congénito)
(consultar Parte sobre Gastrenterologia e Hepatologia)
Anomalias ano-rectais
(ver adiante)
Notas importantes:
|  |
BIBLIOGRAFIA
Ashcraft KW, Holcomb GW III, Murphy JP (eds). Pediatric Surgery. Philadelphia: Saunders, 2005
Bajaj L, Roback MG. Postreduction management of intussusception in a children’s hospital emergency department. Pediatrics 2003; 112: 1302-1307
Baldisseroto M, Maffazzoni DR, Dora MD. Sonographic findings of Meckel´s diverticulitis in children. AJR 2003; 80: 425-428
Basu R, Burge DM. The effect of antenatal diagnosis on the management of small bowel atresia. Pediatr Surg Int 2004; 20: 177-179
Chang Y-J, Chao H-C, Wang C-J, et al. Evaluating pediatric intussusception using 24-hour ultrasound. Paediatrics & Neonatology 2013; 54: 235-238
Coran AG. Pediatric Surgery. Philadelphia: Elsevier, 2013
Cruz M (ed). Tratado de Pediatria. Barcelona: Ergon, 2011
Escobar MA, Ladd AP, Grosfeld JL, et al. Duodenal atresia and stenosis: long term follow-up over 30 years. J Pediatr Surg 2004; 39: 867-871
Fischer TK, Bihrmann K, Perch M, et al. Intussusception in early childhood: a cohort study of 1.7 million children. Pediatris 2004; 114: 782-785
Garcia JJ, Cruz O, Mintegi S, Moreno JM (eds). M Cruz Manual de Pediatria. Madrid: Ergon, 2020
Guimarães JC, Carneiro MJ, Loio P, Macedo A, Tuna ML, et al. Manual Prático de Neonatologia. Lisboa: Hospital de S. Francisco Xavier/Uriage, 2016
Henry MCW, Brever CK, Tashjian DB, et al.The appendix sign: a radiographic marker for irreducible intussusception. J Pediatr Surg 2006; 41: 487-489
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
McInerny T(ed). Tratado de Pediatria /American Academy of Pediatrics. Madrid: Panamericana, 2010
McIntosh N, Helms P, Smyth R, Logan S (eds). Forfar and Arneil´s Textbook of Pediatrics. London: Churchill Livingstone, 2008
Millar AJ, Rode H, Cywes S. Malrotation and volvulus in infancy and childhood. Semin Pediatr Surg 2003; 12: 229-236
Oldham KT, Colombani PM, Foglia RP (eds). Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott Williams & Wilkins, 2005
O´Neill Jr JA, Rowe MI, Grosfeld JL, et al (eds). Pediatric Surgery: Philadelphia: Elsevier, 2017
Polin RA, Lorenz JM. Neonatology. New York: Cambridge University Press, 2008
Polin RA, Abman SH, Rowitch DH, Benitz WE, Fox WW (eds). Fetal and Neonatal Physiology. Philadelphia: Elsevier, 2017
Stringer MD, Spitz L, Abel R. Management of alimentary tract duplication in children. Br J Surg 1995; 82: 74-76
Wyllie R, Hyams JS, Kay M (eds). Pediatric Gastrointestinal and Liver Disease. Philadelphia: Elsevier, 2016
Ziegler M. Meconium ileus. Curr Probl Surg 1994; 34: 731-735