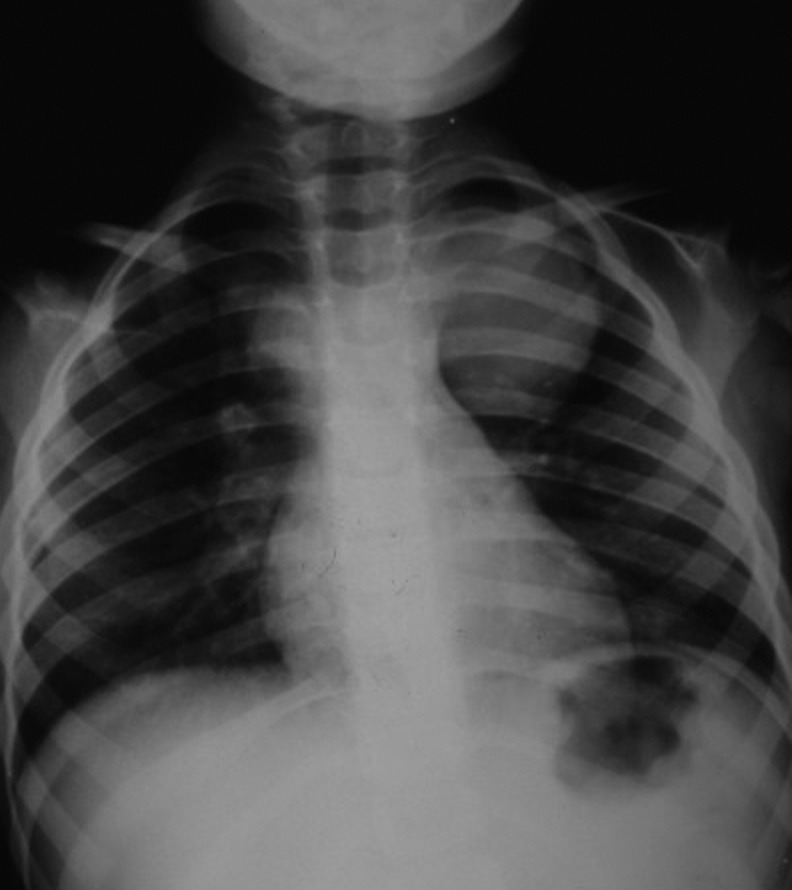
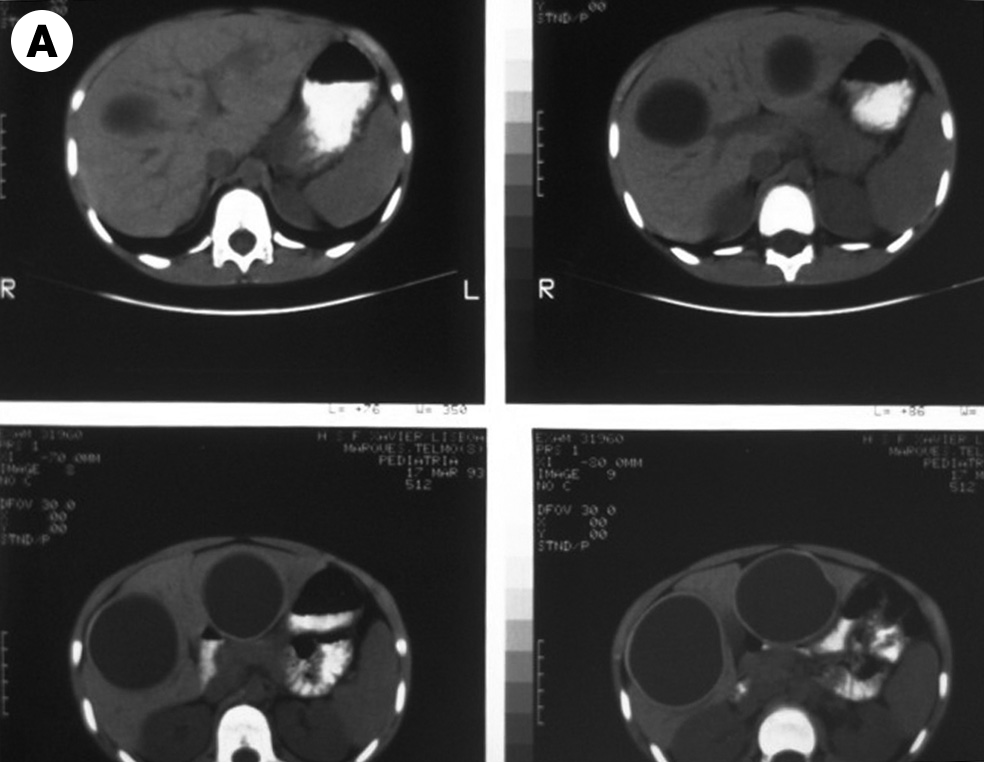
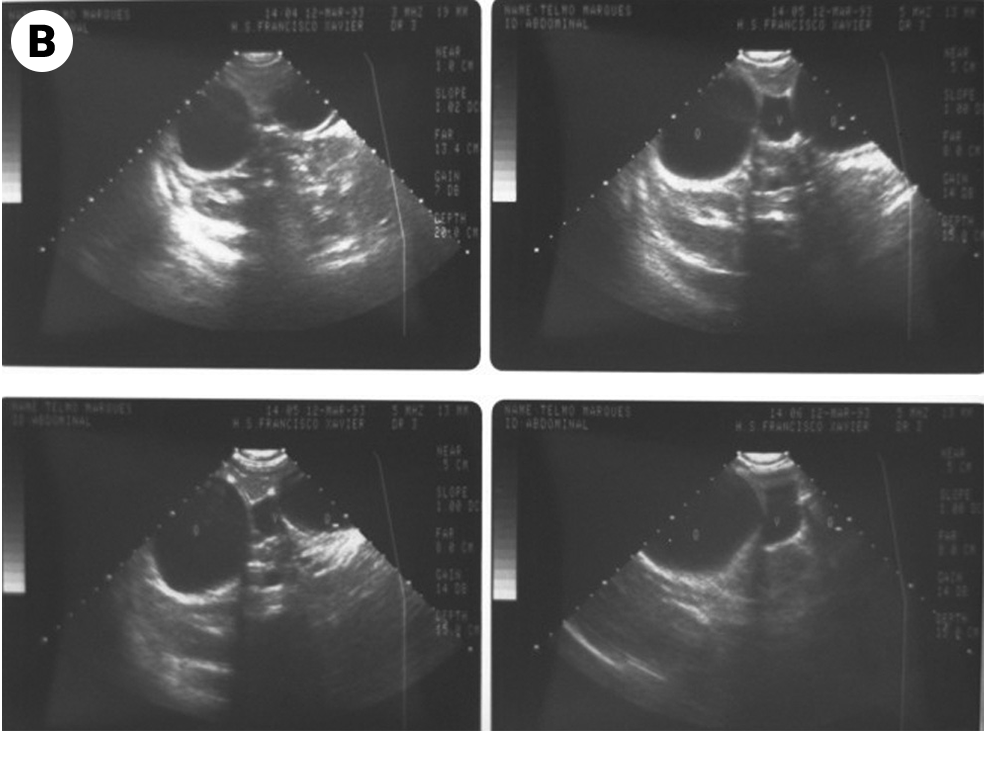
Definições e importância do problema
A Organização Mundial de Saúde considera urgência toda a situação em que, na opinião do doente ou dos seus responsáveis, família ou outra entidade, são requeridos cuidados médicos imediatos. A Comissão Americana para a Medicina de Emergência Pediátrica definiu emergência sob o ponto de vista do utente (prudent-layperson laws): “todo o problema clínico de aparecimento súbito que se manifesta por sintomas suficientemente graves, incluindo dor de grande intensidade, para a qual o leigo prudente que possua conhecimentos médios sobre saúde e medicina, poderá com grande probabilidade esperar que, na ausência de avaliação médica, exista risco para a saúde da pessoa, ou perturbação grave de funções de órgão ou parte do corpo”. Há um grande componente subjectivo nestes conceitos, e uma situação considerada subjectivamente como urgência poderá vir a revelar-se como verdadeira urgência vital ou emergência, susceptível de assistência em serviços com características diversas, ou como não vital – a maioria. (Figura 1)

FIGURA 1. Urgência avaliada inicialmente segundo critérios subjectivos e evolução possível
No nosso País, a Comissão Técnica de Apoio ao Processo de Requalificação das Urgências (2007), estabeleceu as seguintes definições:
- Urgências – todas as situações clínicas de instalação súbita em que existe o risco de falência de funções vitais;
- Emergências – todas as situações clínicas de estabelecimento súbito em que existe, estabelecido ou iminente, o compromisso de uma ou mais funções vitais.
Sistema de cuidados de urgência e emergência
A filosofia de prestação de cuidados baseia-se num sistema que regula relações de complementaridade e de apoio técnico entre instituições hospitalares e não hospitalares, com graus de diferenciação diversos de modo a garantir o acesso atempado de todos os doentes aos serviços e unidades prestadoras de cuidados de saúde em função da patologia detectada. Estes sistemas deverão articular-se entre si explorando complementaridades e concentrando recursos humanos e técnicos, tendo em vista as necessidades reais das populações e a eficiência dos cuidados prestados.
Os utilizadores dos serviços de urgência têm características que os distinguem dos de outros serviços hospitalares:
- A sua chegada não tem marcação;
- A variabilidade das queixas e da gravidade é ampla;
- O cuidado prestado é episódico;
- O recurso ao serviço é muitas vezes inadequado.
De acordo com diversas estatísticas em contextos diversos, entre 20 e 80% das visitas de pacientes aos Serviços de Urgência (SU) pediátricos são motivadas por situações não urgentes. O seu atendimento deveria ter lugar nas instituições devotadas aos cuidados de saúde primários (CSP), o que não sobrecarregaria os SU hospitalares.
Uma vez que, dum modo geral, o cidadão comum e famílias não possuem conhecimentos sobre a orgânica e funcionamento dos serviços de saúde, surgem consequências dramáticas na organização e sustentabilidade dos SU na “pura e técnica” concepção do termo.
A Medicina de Urgência/Emergência, constituindo-se como paradigma actual para a resposta a essas necessidades, inclui diversas vertentes:
- Triagem;
- Avaliação médica de acordo com prioridade;
- Prestação de cuidados (urgentes e emergentes em função do contexto clínico, incluindo lesões traumáticas);
- Encaminhamento dos doentes para seguimento;
- Transporte do doente grave ou com necessidades de cuidados específicos;
- Formação própria;
- Investigação básica e aplicada em aspectos clínicos, mas também de gestão e organização de recursos.
Sobre as vertentes Triagem e Transporte de doentes, as mesmas serão retomadas adiante.
Legislação sobre o Sistema Integrado de Emergência
O despacho nº 10319/2014 sobre o Sistema Integrado de Emergência Médica, que é omisso em orientações para o atendimento e seguimento de situações verdadeiramente não urgentes nos CSP, define a Rede de SU, respectivas responsabilidades e localizações da seguinte forma:
Serviços de Urgência Básica (SUB)
“O atendimento a crianças é da responsabilidade de Médicos e de Enfermeiros não diferenciados em Pediatria, os quais devem receber formação de modo a garantir as competências adequadas ao reconhecimento e abordagem de situações de doença grave, paragem cardíaca, abordagem da via aérea com adjuvantes, acesso vascular emergente e reconhecimento e abordagem inicial da paragem cardíaca em crianças.”
Em todos os SUB deve existir equipamento adequado às diferentes idades pediátricas, para utilização na abordagem correcta da via aérea básica e avançada, na obtenção de acesso vascular urgente e na monitorização em situações de doença grave ou paragem cardíaca; devem dispor de uma sala dedicada ao atendimento de crianças, e que permita, se necessário, a sua permanência para observação de curta duração em espaço separado do atendimento dos adultos. É igualmente considerada desejável a existência de espaços de admissão e salas de espera dedicados à idade.”
Serviços de Urgência Médico-Cirúrgica (SUMC) e Serviços de Urgência Polivalente (SUP)
O atendimento a crianças, da responsabilidade de Serviços de Urgências Pediátricas, deve ter instalações autónomas, incluindo admissão e áreas de espera.
Devem existir, nestas urgências, áreas adequadas a funcionar como salas de observação ou internamento de curta duração (incluindo o de foro ortopédico, cirúrgico e de outras especialidades de apoio). O atendimento deve abranger todos os pacientes em idade pediátrica independentemente da patologia apresentada, excepto nas situações inerentes ou consequentes à gravidez, as quais devem ser atendidas nos serviços de urgência obstétrica.
As Urgências Pediátricas devem funcionar como primeiro ponto de atendimento pediátrico especializado em situações urgentes e emergentes, com base numa lógica de proximidade e organização regional. Devem estar dotadas de canais de comunicação, ágeis e disponíveis, com os SUB e CSP da área, bem como com os serviços para os quais referenciam, nomeadamente Unidades de Cuidados Intensivos Pediátricos e outras áreas de especialidade, tais como Cirurgia Pediátrica, Neurocirurgia e outras. A referenciação para estas Unidades, via transporte inter-hospitalar pediátrico, deve ser protocolada regionalmente e coordenada pelo Instituto Nacional de Emergência Médica (INEM).
As urgências pediátricas de hospitais com SUMC ou SUP devem dispor da presença física permanente de, pelo menos, dois pediatras, um dos quais com formação em suporte avançado de vida pediátrico. Nos hospitais com SUMC, as crianças e jovens com patologia cirúrgica devem ser observados pelos especialistas que prestam cuidados na urgência de adultos, devendo ser protocolada a referenciação de situações clínicas que devam ser transferidas para um SUP Pediátrico.
Para além da disponibilidade dos meios complementares de diagnóstico e terapêutica definidos para os SUMC ou SUP, as Urgências Pediátricas devem dispor de todos os equipamentos específicos da idade pediátrica necessários à abordagem avançada do paciente gravemente doente, traumatizado ou em paragem cardíaca.”
Serviços de Urgência Polivalente Pediátrica (SUPP)
O SUPP deve dispor de todos os recursos mínimos definidos para um SUP, e apoio em termos de diagnóstico e terapêutica e das diversas especialidades, incluindo Neurocirurgia, de Cirurgia Pediátrica e Cuidados Intensivos Pediátricos, local e permanente.
As equipas devem ainda ter formação adequada para que os SUPP funcionem como Centro de Trauma Pediátrico (CTP), devendo o SUPP estar preparado para o atendimento diferenciado de trauma grave, incluindo neurotrauma. O SUPP deve dispor de apoio fácil local ou com garantia de apoio efectivo das áreas de Cardiologia Pediátrica e Pedopsiquiatria. Tal apoio pressupõe a existência de normas rígidas exequíveis.
Triagem
A grande afluência de doentes aos SU em todos os países obrigou ao aperfeiçoamento das normas do atendimento, estabelecendo prioridades em função da gravidade. A finalidade última da triagem é prestar globalmente melhor serviço à comunidade, com rapidez, eficácia e eficiência proporcionais à gravidade. Para evitar iniquidades, estabeleceram-se critérios objectivos (de aplicabilidade, reprodutibilidade e validade), internacionalmente reconhecidos e já utilizados nos sistemas de triagem estruturados noutros Países.
Estes sistemas pressupõem obrigatoriamente os seguintes requisitos:
- Definição de 5 níveis de prioridade (gravidade);
- Definição de tempos máximos de espera aguardando observação médica de cada caso clínico previamente analisado;
- Auditoria realizada por entidades externas.
A triagem é um processo dinâmico e exigente que se inicia quando o paciente chega ao serviço de urgência, e finaliza quando este recebe uma avaliação completa por um médico. Neste processo, requer-se, não só a capacidade de reconhecer os sinais e sintomas que necessitam de ser tratados imediatamente, mas também o reconhecimento de sintomas que provavelmente corresponderão a uma doença benigna.
Durante o tempo de espera os pacientes podem melhorar ou piorar; por isso, torna-se necessário proceder a reavaliações periódicas (retriagem).
Nesta perspectiva, a triagem implica, pois, formação e aperfeiçoamento dos profissionais que a realizam, estando bem definidas as características e as responsabilidades de tal função.
Os sistemas de triagem recomendados para a Pediatria em Portugal são o Manchester Triage Scale (MTS) e o Canadian Paediatric Triage and Acuity Scale (CPTAS). (Quadro 1)
QUADRO 1 – Sistemas internacionais de triagem pediátrica em Portugal
| MTS | CPTAS | |
| Nível de Gravidade (sinalização com cor) | Tempo de Resposta Médica Alvo (minutos) | Tempo de Resposta Médica Alvo (minutos) |
| 1 = Imediata (Vermelho) 2 = Muito Urgente (Laranja) 3 = Urgente (Amarelo) 4 = Menos Urgente (Verde) 5 = Não Urgente (Azul) | Imediato 10 60 120 240 | Imediato 15 30 60 120 |
Transporte de doentes
Os sistemas de transporte pediátrico e neonatal inter-hospitalar permitem que os doentes beneficiem de cuidados especializados antes e durante a transferência. Está demonstrado em diversos estudos que o transporte, incorporando equipa médica e de enfermagem especializadas, permite reduzir a mortalidade e morbilidade, verificando-se também uma boa relação custo-benefício.
Existem diversos modelos de organização de sistemas de transporte, nem sempre consensuais. O ideal será, pois, criar condições para que o sistema de transporte se desloque ao local onde existe um doente em estado crítico requerendo tratamento emergente, e não o contrário.
Reproduzindo o que foi estabelecido oficialmente, importa realçar certos princípios: “Em Portugal o transporte pré-hospitalar é assegurado pelo INEM, dependente do Ministério da Saúde.
Quanto ao transporte inter-hospitalar neonatal e pediátrico destacam-se principalmente três períodos:
- A experiência nacional desde 1978, com o transporte inter-hospitalar especializado de recém-nascidos (RN), de cobertura nacional, no âmbito do INEM;
- A experiência da região centro do país desde 1991 coordenada a partir do Hospital Pediátrico de Coimbra, também no âmbito do INEM; tal subsistema assegura, não só o transporte de RN de alto risco, mas igualmente o de doentes pediátricos necessitando de cuidados intensivos;
- O modelo actual, a funcionar desde 2013, em que houve uma uniformização e integração a nível nacional da gestão altamente diferenciada do Transporte Inter-hospitalar Pediátrico (TIP); tal modelo tem como missão:
- a deslocação rápida de uma equipa de transporte de doente crítico urgente em idade pediátrica;
- a estabilização clínica dos recém-nascidos e/ou crianças gravemente doentes; e
- o transporte acompanhado para unidades de cuidados intensivos neonatais e/ou pediátricas disponíveis.”
Reforçando o que foi abordado anteriormente, o sistema de transporte implica igualmente o estabelecimento de normas de actuação médica e organizativa, assim como recursos logísticos tais como: equipa médica e de enfermagem treinada autónoma, meios de transporte por via terrestre ou aérea, aparelhagem específica, oxigénio e ar armazenados com possibilidade de ventilação mecânica, fármacos, etc.. (Quadros 2 e 3)
QUADRO 2 – Equipamento indispensável durante o transporte
|
QUADRO 3 – Fármacos e fluidos indispensáveis durante o transporte
|
Uma norma basilar aplicável ao transporte de doentes em qualquer grupo etário diz respeito à garantia de estabilização hemodinâmica, antes de se iniciar o transporte, e à ponderação dos benefícios face aos riscos.
Com efeito, o hospital de proximidade da ocorrência, necessitando de transferência de doentes/hospital “emissor” (por doença ou por lesão traumática), deve ter:
- Capacidade para a estabilização do doente antecedendo uma transferência;
- Plano de transferências e transportes que permita enviar de modo sistemático, em segurança e atempadamente, um doente para um centro de maior diferenciação/hospital “receptor”, pré-identificado, que proporcione cuidados definitivos.
Unidades de cuidados intensivos pediátricos (UCIP)
Uma parcela limitada dos doentes recorrendo aos SU/E abertos ao exterior, ou transferidos doutros hospitais, requerem cuidados designados por intensivos pela situação clínica considerada crítica.
Considera-se doente crítico aquele em que, por disfunção ou falência profunda de um ou mais órgãos ou sistemas, a sobrevivência depende de:
- Recursos humanos altamente especializados integrando equipas próprias médico-cirúrgicas e de enfermagem altamente na relação de 1 enfermeira/doente (permanentes, 7 dias por semana, 24 horas por dia, 365 dias por ano), e o apoio de múltiplos subespecialistas;
- Meios sofisticados de terapêutica (por ex.: ventilação mecânica, hemodiálise, circulação extracorporal, farmacoterapia complexa, terapia pós-transplantes, etc.); e
- Diversos tipos de monitorização contínua (electrónica, biofísica, bioquímica/laboratorial, invasiva e não invasiva, etc.).
Pelos elevados custos que tal tipo de cuidados exige, e pela necessidade de ser criada massa crítica com vista à aquisição de experiência e aperfeiçoamento de competências por parte das equipas assistenciais, garantindo a qualidade dos mesmos cuidados, torna-se necessário concentrar recursos humanos e materiais nas chamadas unidades de cuidados intensivos (neste caso pediátricos, com número limitado de postos), localizadas em hospitais do mais elevado nível de diferenciação na prestação de cuidados hospitalares (nível terciário), com esquemas organizativos variáveis, a que atrás se aludiu.
Urgências e Emergências Pediátricas – o presente e o futuro
Nas décadas recentes ocorreram grandes avanços no âmbito da prestação e organização de cuidados pediátricos urgentes e emergentes a nível mundial, com maior relevância nos países ditos desenvolvidos. Com efeito, de acordo com a experiência acumulada, concluiu-se que se torna indispensável considerar a valência Urgências Pediátricas como uma subespecialidade pediátrica, implicando a criação de equipas (designadamente médicas e de enfermagem), com sólida formação global e com competências específicas (designadamente técnicas) para o tratamento de doentes complexos em estado crítico.
Estas equipas participariam, não só na assistência médica directa, mas também noutras tarefas: auditorias internas devotadas ao atingimento de metas de qualidade assistencial dos respectivos serviços; consultas de reavaliação de situações agudas mais complexas; consultas sem presença de doente; ligação a instituições (de proximidade, emissoras de pacientes como anteriormente referido, quer hospitais, quer centros de saúde); acções de formação, incluindo as relacionadas com treino em simulação de técnicas; criação e discussão de normas de orientação clínica; investigação, designadamente no âmbito da comunicação médico-paciente e interpares, analgesia, sedação, avaliação do risco clínico e técnicas de imagiologia rápida como a ecografia, etc..
BIBLIOGRAFIA
Abecasis F. Transporte neonatal e pediátrico. Organização e perspectivas actuais. Nascer e Crescer 2008; 17: 162-165
Ajizian SJ, Nakagawa TA. Interfacility transport of the critically ill pediatric patient. Chest 2007; 132: 1361-1367
American Academy of Pediatrics. Section on Transport Medicine. Guidelines for air and Ground Transport of Neonatal and Pediatric Patients. Elk Grove Village, IL: AAP, 2007
American Academy of Pediatrics, Committee on Pediatric Emergency Medicine Services for Children in Emergency Departments. Elk Grove Village, IL: AAP ed, 2007
Ames SG, Davis BS, Marin JR, et al. Emergency department pediatric readiness and mortality in critically Ill children. Pediatrics Sep 2019; 144: (3) e20190568; DOI: 10.1542/peds.2019-0568
Andrews B, Pinto N. Optimizing discharge from intensive care and follow-up strategies for pediatric patients. J Pediatr 2019; 205: 8
Baren JM, Rothrock SG, Brennan (eds). Pediatric Emergency Medicine. Philadelphia: Lance Brown/ Saunders, 2007
Benito X, Mintegi S, Ruddi RM, Del Rey JG. Changing clinical practices and education in pediatric emergency medicine through global health partnerships. Clin Pediatr Emergency Med 2012; 13: 37-43
Carvalho IP, et al. Recomendação de um Modelo Nacional de Triagem Pediátrica. Proposta. Comissão Nacional da Saúde Materna, da Criança e do Adolescente. Julho. 2013. www.dgs.pt/directrizes-da…/norma-n-0022015-de-06032015-pdf.aspx ( acedido em 9 de Maio de 2017)
Chen C, Scheffler G, Chandra A. Massachusetts’ health care reform and emergency department utilization. N Engl J Med 2011; 365: e25
Comissão Nacional de Saúde da Criança e Adolescente. Carta Hospitalar de Pediatria (documento em discussão). Lisboa: Ministério da Saúde ed, 2008
Cozzi G, Barbi E. Facing somatic symptom disorder in the emergency department. J Paediatr Child Health 2019; 55: 7-9
Cruz AT. Indications and interpretation of common laboratory assays in the Emergency Department. Pediatr Clin North Am 2018; 65: 1191-1204
Despacho nº5058-D/2016 publicado no Diário da República nº 72, 2ª série, de 13 Abril
Direcção Geral da Saúde, Norma 002/2015 de 23 de Outubro “Sistemas de Triagem dos Serviços de Urgência e Referenciação Interna Imediata”. Lisboa: DGS, 2015
Escobedo MB, Shah BA, Song S, et al. Recent recommendations and emerging science in neonatal resuscitation. Pediatr Clin N Am 2019; 66: 309-320
Fuhrman BP, Zimmerman J (eds). Pediatric Critical Care. Philadelphia: Mosby, 2006
Goldman L, Schafer AI (eds). Goldman – Cecil Medicine. Philadelphia: Elsevier Saunders, 2016
Grupo Português de Triagem de Prioridades na Urgência. Declaração de Princípios. Lisboa: Ministério da Saúde, 2008
Helfaer MA, Nichols DG. Roger’s Handbook of Pediatric Intensive Care. Philadelphia: Lippincott Williams & Wilkins, 2009
Januário L. Urgência pediátrica: um serviço do hospital em movimento com cérebro. (Editorial) Acta Pediatr Port 2016; 47: I
Kliegman RM, StGeme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds). Nelson Textbook of Pediatrics. Philadelphia: Elsevier, 2020
Kline MW, Blaney SM, Giardino AP, Orange JS, Penny DJ, Schutze GE, Shekerdemien LS (eds). Rudolph’s Pediatrics. New York: Mc Graw Hill Education, 2018
Ku BC, Chamberlain JM, Shaw KN. Quality improvement and safety in Pediatric Emergency Medicine. Pediatr Clin North Am 2018; 65: 1269-1282
Lee JH, Choong K. Time to focus on paediatric critical care survivorship. Lancet Respir Med 2019; 7: 103-105
Louie MC, Chang TP, Grundmeier RW. Recent advances in technology and its applications to Pediatric Emergency Care. Pediatr Clin North Am 2018; 65: 1229- 1237
Medford-Davis LN, Singh H, Mahajan P. Diagnostic decision-making in the Emergency Department. Pediatr Clin North Am 2018; 65: 1097-1106
Ministério da Saúde. Despacho nº 10319/2014 de 11 de Agosto. ”Estrutura do sistema integrado de emergência médica (SIEM)”. Lisboa: DGS, 2014
Moro M, Málaga S, Madero L (eds). Cruz Tratado de Pediatria. Madrid: Panamericana, 2015
Pitts SR, Carrier ER, Rich EC, et al. Where Americans get acute care: increasingly, it’s not at their doctor’s office. Health Aff (Millwood) 2010; 29: 1620-1629
Polin RA, Yoder MC. Workbook in Practical Neonatology. Philadelphia: Elsevier Saunders, 2015
Rae P, Jenks S. Optimal use of blood tests in acute medicine. Medicine 2013; 41: 132- 134
Rosen JR, Suresh S, Saladino RA. Quality care and patient safety in the pediatric emergency department. Pediatr Clin North Am 2016; 63: 269-282
Royal College of Paediatric and Child Settings; (2012) Standards for children and young people in emergency care settings. http://www.rcpch.ac.uk/sites/default/files/page/Intercollegiate Emergency Standards (acesso em Dezembro de 2019)
Sankrithi U, Schor J. Pediatric urgent care – New and evolving paradigms of acute care. Pediatr Clin North Am 2018; 65: 1257-1268
Shenoi RP, Timm N, AAP Committee on Drugs. AAP Committee on Pediatric Emergency Medicine. Drugs used to treat pediatric emergencies. Pediatrics 2020; 145(1): e2019345
Smith KA, Flori HR. Critical care in the pediatric emergency department. Pediatr Clin North Am 2018; 65: 1119-1134
Sociedade Portuguesa de Cuidados intensivos/Ordem dos Médicos. Transporte de Doentes Críticos. Recomendações. Lisboa: Ordem dos Médicos ed, 2008
Vaughan L. Biomarkers in acute medicine. Medicine 2013; 41: 136-141